
Abstract
Long non-coding RNAs recently were identified as key mediators of cancer metastasis. This study provided evidence that long non-coding RNA MALAT1 was up-regulated in breast cancer tissues and cell lines. MALAT1 promoted cancer cell invasion through inducing epithelial–mesenchymal transition. Interestingly, we revealed there was a reciprocal repression between MALAT1 and miR-204. ZEB2 was identified as a downstream target of miR-204 and MALAT1 exerted its function mainly through the miR-204/ZEB2 axis. Our findings suggested that MALAT1 may serve as a new diagnostic biomarker and therapy target for breast cancer.
Introduction
Breast cancer is a common malignancy around the world, with an incidence of 370,000 deaths annually. Invasion and metastasis are significantly aggressive phenotypes of breast cancer. Literatures suggest that epithelial–mesenchymal transition (EMT) is a critical mechanism that contributes to the invasion and metastasis of breast cancer. 1 To understand the mechanism of invasion and metastasis and develop novel therapy strategy, identification of key inducers of EMT is of critical importance.
MicroRNAs (miRNAs) are small non-coding RNAs of about 22 nucleotides (nt) in length that are found in all eukaryotic cells. It is well documented that miRNAs are involved in diverse biological processes, including differentiation, proliferation, apoptosis, and tumorigenesis. Recently, it is documented that miRNAs may potentially interact with long non-coding RNA (lncRNA) and modulate each other’s expression.2,3
Unlike the miRNAs, lncRNAs are by definition >200 nt in length. A growing number of literature suggests that lncRNAs play an important role in cancer progress.4,5 Interestingly, lncRNAs and miRNAs could interact with each other, imposing an additional level of post-transcriptional regulation.6,7 For instance, lncRNA GAS5 could bind at miRNA-21 by acting as a negative regulator of miR-21. 6
In this study, we investigated the role of lncRNA MALAT1 on promoting EMT and its interaction with miR-204 in breast cancer.
Materials and methods
Breast cancer cell culture and tissue samples
Breast cancer cell lines and normal immortalized MCF-10A cells were obtained from Key Laboratory of Malignant Tumor Gene at Sun Yat-sen University. The cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS). All cell lines were maintained at 37°C in a humidified atmosphere with 5% CO2.
Breast cancer patients those diagnosed by histopathology in the Affiliated Hospital of Guangdong Medical University were obtained. This study was conducted with the approval of the Ethical and Scientific Committees of Guangdong Medical University with its number GZM-2015-MR8.
Quantitative reverse transcription–polymerase chain reaction assay
Total RNA was isolated from the breast cancer patient tissues and breast cancer cell lines using Trizol reagent. cDNA from total RNA was generated using a high-capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). cDNA from miRNAs was then generated using TaqMan miRNA Reverse Transcription kit (Applied Biosystems). Quantitative reverse transcription–polymerase chain reaction (qRT-PCR) was conducted using TaqMan gene expression assays of MALAT1 and GAPDH or TaqMan Universal Master Mix II with TaqMan microRNA assays of miR-204 and U6. The primers are listed in Table 1.
The primers used in this study.

In situ hybridization
Five-micrometer-thick formalin-fixed paraffin-embedded tissue sections were hybridized with the MALAT1 probe (LNA-modified and 5′- and 3′- DIG-labeled oligonucleotide; Exiqon, Woburn, MA). The subsequent procedure was carried out as previously described. 8
Transwell and Boyden assays
For transwell assay, cells in 100-mL PRMI-1640 medium without FBS were seeded on a fibronectin-coated polycarbonate membrane insert in a transwell apparatus. In the lower chamber, 600-mL PRMI-1640 with 10% FBS was added as a chemoattractant. Then, the cells were incubated for 8 h at 37 uC in a 5% CO2 atmosphere. Subsequently, the insert was washed with PBS; cells adhering to the lower surface were fixed with methanol, stained with Giemsa solution, and counted.
Western blot and immunofluorescence assays
For western blot assay, equal amounts of protein were resolved by SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membrane. Then, the membranes were blocked in 5% non-fat skim milk/TBST, followed by detection of primary antibodies at 4°C overnight. Subsequently, the membranes were detected with appropriate secondary antibodies. The levels of goal protein were detected with enhanced chemiluminescence reagents (Pierce, Rockford, IL). The primary antibodies E-cadherin (Lot no. sc-7870), N-cadherin n (Lot no. sc-393933), Vimentin (Lot no. sc-5565), ZEB2 (Lot no. sc-48789), and GAPDH (Lot no. sc-48166) were purchased from Santa Cruz (USA). The second antibodies IgG(H+L)(HRP-labeled Goat Anti-Rabbit IgG(H+L)) and IgG(H+L)(HRP-labeled Rabbit Anti-mouse IgG(H+L)) were purchased from ZSGB-BIO (Beijing, China). The immunofluorescence assay was performed as previously described. 9
Oligonucleotide transfection
Synthesized RNA duplexes of miRNA control, miR-204, and anti-miR-204 were obtained from Ribobo (Guangzhou, China). pcDNA3-ZEB2 was purchased from Biosettia Inc. (San Diego, CA). Oligonucleotide transfection was performed with Lipofectamine 2000 reagent (Invitrogen; Thermo Fisher Scientific corporation, California, USA).
Luciferase reporter assay
The 3′-UTR untranslated region of ZEB2 was amplified by PCR and cloned downstream of the firefly luciferase gene in the pGL3 vector (Promega). The vector was named ZEB2-3′-UTR. Cells were tranfected with ZEB2-3′-UTR and miR-ctrl or miR-204. The luciferase assay was performed using the dual Luciferase reporter assay system (Promega; Fitchburg, Wisconsin, USA) 48 h after transfection.
Lentivirus production and transduction
Lentiviral shRNA targeting MALAT1 was designed at http://biosettia.com/support/shrna-designer and cloned into pLV-H1TetO-GFP-Puro vector according to manufacturer’s instructions (Biosettia). The viruses were packaged in 293T cells according to standard protocols, and the virus particles were harvested 72 h later. The packaged lentiviruses were named sh-MALAT1 and the empty lentiviral vector sh-ctrl was used as a control. Cells were infected with virus particles plus 8 µg/mL polybrene.
In vivo study
The experimental metastasis studies were conducted as previously described. 10 The cells were injected into the mice via the tail vein. Five weeks later, the mice were sacrificed and lung metastases were detected using haematoxylin and eosin (H&E) staining.
Statistical analysis
SPSS software was used for statistical analysis. Data are represented as mean ± standard error of mean (SEM). Fisher’s exact test was used to identify differences between categorical variables. One-way ANOVA or two-tailed Student’s t-test was used for comparisons between groups. Differences were considered statistically significant when p < 0.05.
Results
MALAT1 was up-regulated in breast cancer tissues and cell lines
We first examined the expression of MALAT1 in 118 human breast cancer tissues and adjacent normal breast tissues by RT-PCR. It was found that MALAT1 expression was significantly up-regulated in breast cancer tissues when compared with normal breast tissues (Figure 1(a)). In addition, the expression of MALAT1 was elevated in breast cancer cell lines when compared with normal immortalized MCF-10A cells (Figure 1(b)).

MALAT1 was up-regulated in breast cancer tissues and cell lines. (a) MALAT1 expression was significantly up-regulated in breast cancer tissues when compared with normal breast tissues. (b) The expression of MALAT1 was elevated in breast cancer cell lines when compared with normal immortalized MCF-10A cells. (c) High expression of MALAT1 was frequently observed in M1, and III-IV tumors than M0, and I-II tumors. (d) Patients with higher levels of MALAT1 expression had poorer overall survival than those with lower levels of MALAT1 expression.
MALAT1 expression was associated with aggressive and poor prognostic of breast cancer
We next explored the relation between MALAT1 expression and clinicopathologic characteristics of breast cancer patients. Although MALAT1 expression was not associated with age, gender, ER status, PR status, and tumor size (T classification), MALAT1 expression was associated with metastasis (M classification) (p = 0.001) and clinical stage (p = 0.026) (Table 2). High expression of MALAT1 was frequently observed in M1, and III-IV tumors than M0, and I-II tumors (Figure 1(c)). These data suggested that MALAT1 expression may be involved in metastasis and invasion of breast cancer. Importantly, patients with higher levels of MALAT1 expression had poorer overall survival than those with lower levels of MALAT1 expression (Figure 1(d)).
Relation between MALAT1 expression and clinicopathologic characteristics of breast cancer patients.
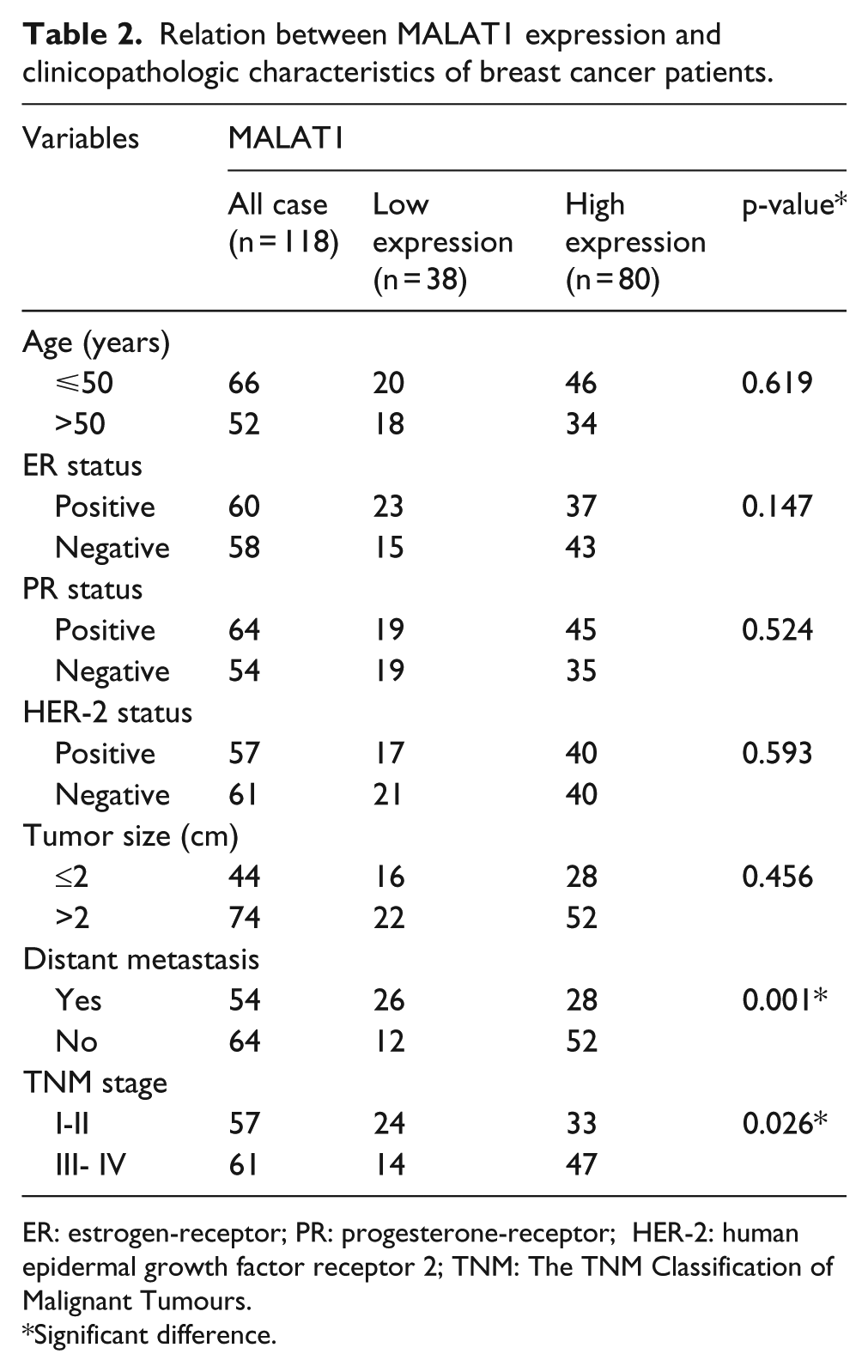
ER: estrogen-receptor; PR: progesterone-receptor; HER-2: human epidermal growth factor receptor 2; TNM: The TNM Classification of Malignant Tumours.
Significant difference.
MALAT1 down-regulation inhibited cell motility and invasion through reversed EMT phenotype
We first established MCF-7 cells that stably expressed low MALAT1 and named them shMALAT1 cells. The cells used for negative control were named sh-ctrl cells. It was revealed that MALAT1 down-regulation inhibited breast cancer cell migration and invasion, as determined by transwell and Boyden assay (Figure 2(a) and (b)). EMT is a central mechanism contributing to invasion and metastasis of breast cancer. We thus asked whether MALAT1 down-regulation reversed EMT phenotype. The western blot assay revealed that MALAT1 down-regulation increased epithelial markers E-cadherin while decreasing mesenchymal markers Vimentin and N-cadherin (Figure 2(c)). Similarly, the immunofluorescence assay demonstrated that MALAT1 down-regulation could increase epithelial marker but decrease mesenchymal markers (Figure 2(d)).

MALAT1 down-regulation inhibited cell motility and invasion through reversed EMT phenotype. (a) The transwell assay revealed that MALAT1 down-regulation inhibited breast cancer cell migration. (b) The Boyden assay showed that knockdown of MALAT1 decreased invasion ability of breast cancer cell (c) and (d) the western blot and immunofluorescence assays revealed that MALAT1 down-regulation increased epithelial markers E-cadherin while decreasing mesenchymal markers.
The interaction between MALAT1 and miR-204
The online software program starBase v2.0 (http://starbase.sysu.edu.cn/mirLncRNA.php) 11 was used to search for miRNAs that have complementary base pairing with MALAT1. Among the predicated microRNAs that may interact with MALAT1, we focused on miR-204 since miR-204 inhibited cell invasion through reversing EMT in various cancers, including breast cancer.1,12,13
Luciferase reporter constructs were generated to confirm the direct binding between MALAT1 and miR-204. The wild-type (wt) MALAT1 and mutant (mut) MALAT1 are shown in Figure 3(a). We observed that miR-204 reduced the luciferase activities of wt MALAT1 reporter vector instead of mut MALAT1 reporter (Figure 3(b)). These data suggested that the binding sites are vital for the reciprocal repression of MALAT1 and miR-204.

The interaction between MALAT1 and miR-204. (a) The biding sites of miR-204 on wild-type MALAT1 (indicated in red) and the mutant-type MALAT1 (indicated in blue). (b) MiR-204 mimic reduced the luciferase activities of wt MALAT1 reporter vector instead of mut MALAT1 reporter. (c) Down-regulation of MALAT1 increased miR-204 expression in MCF-7 cells. (d) MiR-204 inhibited MALAT1 expression, whereas miR-204 inhibitor increased MALAT1 expression.
We further revealed that down-regulation of MALAT1 increased miR-204 expression in MCF-7 cells (Figure 3(c)). However, miR-204 inhibited MALAT1 expression, whereas miR-204 inhibitor increased MALAT1 expression (Figure 3(d)). Taken together, these data suggest that there was a reciprocal repression between miR-204 and MALAT1.
MALAT1-controlled miR-204/ZEB2 axis
ZEB2 was predicated as a downstream target of miR-204 using Targetscan and miRanda. We then asked whether miR-204 negatively regulated ZEB2 expression. ZEB2 wild-type (WT) or mutant 3′-UTR (MT) (Figure 4(a)) was subcloned into a luciferase reporter vector and co-transfected with miR-204 mimic or negative control into MCF-7 cells. We found that miR-204 significantly inhibited the luciferase activity of the ZEB2 WT 3′-UTR instead of the mutant 3′-UTR (Figure 4(b)). Furthermore, it was found that overexpression of miR-204 reduced the ZEB2 mRNA level and protein level (Figure 4(c)).Taken together, these findings suggested that ZEB2 was a direct target of miR-204.

MALAT1 controlled miR-204/ZEB2 axis. (a) The ZEB2 mutant sequences and slug wild-type sequences were indicated. (b) miR-204 significantly inhibited the luciferase activity of the ZEB2 WT 3′-UTR but not that of the mutant. (c) miR-204 reduced the ZEB2 mRNA level and protein level. (d) When in the presence of pCDNA-MALAT1, ZEB2 repression was restored compared with the control group. (e) Knockdown of MALAT1 triggered a significant silencing effect on endogenous ZEB2 in MCF-7 cells. (f) shMALAT1 cells established smaller lung metastatic colonies than the control group.
Subsequently, we tested whether MALAT1 regulated ZEB2 expression. The ZEB2 wild-type plasmid was together transfected with miR-204 mimic and MALAT1 (pCDNA3.1-MALAT1). We revealed that ZEB2 repression was restored when in the presence of pCDNA3.1-MALAT1 (Figure 4(d)). Importantly, MALAT1 down-regulation decreased the endogenous ZEB2 expression, as determined by western blot assay (Figure 4(e)). Interestingly, we found that ZEB2 inhibition could mimic the MALAT1 down-regulation effect in breast cancer cells (Supplement Figure 1).
Inhibition of MALAT1 decreased MCF-7 cells metastasis in vivo
To determine whether MALAT1 down-regulation decreased MCF-7 cells metastasis ability in vivo, we performed experimental metastasis studies. We revealed that shMALAT1 cells established smaller lung metastatic colonies than the control group (Figure 4(f)).
Discussion
LncRNA MALAT1 was often overexpressed in many cancers, including glioblastoma, 14 prostate cancer, 15 and esophageal squamous cell carcinoma, 16 including breast cancer. 17 However, the underlying mechanism of MALAT1 in regulating metastasis and EMT is still poorly understood in breast cancer. In this study, we revealed that MALAT1 expression was up-regulated in breast cancer tissues and cell lines. Patients with higher expression of MALAT1 tend to had poorer overall survival than those with lower expression of MALAT1. Our findings revealed that MALAT1 may be a potential prognostic factor in breast cancer.
Evidence has demonstrated that MALAT1 contributes to cancer invasion and metastasis. For instance, MALAT1 could be served as a predictor of NSCLC brain metastasis and MALAT1 promoted brain metastasis through inducing EMT. 18 The MAPK-ERK signaling pathway was suggested to be involved in MALAT1-induced metastasis. 19 Recently, literatures provided an important clue about microRNA–lncRNA interaction during cancer progression.20,21 In this case, lncRNAs may function as competing endogenous RNAs (ceRNAs) to sponge miRNAs, thereby modulating the derepression of miRNA targets and imposing an additional level of post-transcriptional regulation. 22 Our findings first revealed that there were putative biding sites of miR-204 on MALAT1. MiR-204 mimic decreased MALAT1 expression, whereas miR-204 inhibitor increased MALAT1 expression. However, MALAT1 down-regulation led to increased expression of miR-204. Taken together, we proposed that there was an interaction between MALAT1 and miR-204 in breast cancer.
MiR-204 was suggested to be a tumor suppressor and its expression was frequently down-regulated in cancer. Restoration of miR-204 inhibited cancer cell invasion through regulating EMT phenotype. MiR-204 exerted its function mainly by negatively downstreaming its targets, such as slug, 13 SIRT1, 12 and SMAD4, 23 which were the key regulators of EMT. In this study, we identified ZEB2 as a new target of miR-204. ZEB2 was an important mediator of epithelial dedifferentiation through direct down-regulation of a distinct set of constituents of adherens junctions, tight junctions, desmosomes, and gap junctions, which are the key determinants of the epithelial phenotype, including epithelial cell polarity. 24 ZEB2 could induce EMT through directly inhibiting E-cadherin, including breast cancer. 25 Indeed, overexpression of miR-204 decreased ZEB2 expression, as determined by luciferase assay and western blot assay. Furthermore, we found that MALAT1 down-regulation triggered decreased ZEB2 expression. Based on the above findings, we suggest that MALAT1 negatively regulates miR-204 expression by acting as an endogenous sponge. MiR-204 inhibited ZEB2 expression through binding at ZEB2 3-UTR uncoding region. Thus, MALAT1 regulated the miR-204/ZEB2 axis in breast cancer. Given that ZEB2 is a key factor in EMT; 26 we thus proposed that MALAT1 may increase cell metastasis and induce EMT phenotype through miR-204/ZEB2 axis.
In summary, our findings suggested that MALAT1 may represent a biomarker of poor prognosis of breast cancer. Targeting MALAT1 may represent a novel therapeutic application to breast cancer.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.