
Abstract
Somatic mutation analysis is a standard of practice for human cancers to identify therapeutic sensitization and resistance mutations. We performed a multigene sequencing screen to explore mutational hotspots in cancer-related genes using a semiconductor-based sequencer. DNA from oral squamous cell carcinoma samples was used as a template to amplify 207 regions from 50 cancer-related genes. Of the 80 oral squamous cell carcinoma specimens from Japanese patients, including formalin-fixed paraffin-embedded samples, 56 specimens presented at least one somatic mutation among the 50 investigated genes, and 17 of these samples showed multiple gene somatic mutations. TP53 was the most commonly mutated gene (50.0%), followed by CDKN2A (16.3%), PIK3CA (7.5%), HRAS (5.0%), MET (2.5%), and STK11 (2.5%). In total, 32 cases (40.0%) were human papillomavirus positive and they were significantly less likely to have a TP53, mutation than human papillomavirus–negative oral squamous cell carcinomas (8/32, 25.0% vs 32/48, 66.7%, p = 0.00026). We also detected copy number variations, in which segments of the genome could be duplicated or deleted from the sequencing data. We detected the tumor-specific TP53 mutation in the plasma cell-free DNA from two oral squamous cell carcinoma patients, and after surgery, the test for these mutations became negative. Our approach facilitates the simultaneous high-throughput detection of somatic mutations and copy number variations in oral squamous cell carcinoma samples.
Introduction
Oral, pharyngeal, and laryngeal squamous cell carcinomas are grouped together as head and neck squamous cell carcinoma (HNSCC). Oral squamous cell carcinoma (OSCC) is the most common head and neck tumor and accounts for >90% of malignancies affecting the oral cavity. 1 Although the oral cavity is readily accessible for clinical examination, patients with OSCC often present with symptoms at a late stage, thereby limiting the effectiveness of chemotherapy, radiotherapy, and surgery. Therefore, OSCC continues to have a poor prognosis, with a 5-year survival rate of <50%.2,3 Research directed toward the identification of biomarkers for the early detection of OSCC, indicators of good or bad prognosis, and determinants of treatment response is essential.4,5 However, the precise molecular mechanism underlying the chemotherapy resistance of recurrent OSCC remains largely unknown.
Recent advances in next-generation sequencing (NGS) technologies provide a powerful tool for elucidating the genetic basis of tumor initiation and progression, making this an attractive approach to better guide personalized precision medicine.6,7 Whole-genome sequencing and whole-exome sequencing (WES) are useful methods for detecting somatic mutations in human cancers. However, the routine use of these technologies presents several limitations, such as the high entry cost, long processing time, and sample scalability. Although mostly sufficient for histopathological diagnosis, only very limited amounts of formalin-fixed paraffin-embedded (FFPE) tissue are often available for predictive mutation analysis. In diagnostic clinical settings, targeted NGS has become a widely accepted method for simultaneously interrogating multiple lung cancer–associated genes in non-small cell lung cancers (NSCLCs), 8 but this approach has not yet been reported in OSCC. In addition, few studies to date have focused on the mutation spectrum of OSCC patients in Japan.
The semiconductor sequencing platform, which makes DNA sequencing more reliable, faster, and cost-effective, has a very low input DNA requirement and is compatible with FFPE samples. 9 In this study, 18 OSCC cell lines and surgically resected OSCC tissues from 80 Japanese patients were analyzed for mutations in mutational hotspot regions from 50 cancer-related genes using a semiconductor-based Ion Torrent sequencer. This targeted NGS has significant advantages over classic molecular methods for use in clinical laboratories that perform high-throughput sequencing.
Materials and methods
Barcoded genomic DNA library construction
Amplicon libraries for individual DNA samples were prepared using the Ion AmpliSeq Cancer Hotspot Panel v2 (CHPv2) and the Ion AmpliSeq Library Kit 2.0 (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol without modifications. Multiplex polymerase chain reaction (PCR) was performed in 17 cycles for frozen samples and in 20 cycles for FFPE samples according to the manufacturer’s recommendations. This panel targets 2855 COSMIC mutational hotspot regions in the following 50 genes: ABL1, AKT1, ALK, APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, EZH2, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAS, GNAQ, HNF1A, HRAS, IDH1, IDH2, JAK2, JAK3, KDR, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53, and VHL. Sequencing adapters with unique barcodes (Ion Xpress Barcode Adapters; Thermo Fisher Scientific) were ligated to the amplification products and purified using Agencourt AMPure XP Reagent (Beckman Coulter, Brea, CA, USA) according to the manufacturer’s instructions.
Semiconductor-based NGS using the Ion Torrent platform
The libraries were quantified using a real-time PCR approach and the Ion Library Quantitation Kit (Thermo Fisher Scientific). The assessment of library quality (molarity and size analysis) based on FFPE samples was carried out using an Agilent 2100 Bioanalyzer and the Agilent High Sensitivity DNA Kit (Agilent Technologies, Santa Clara, CA, USA). A total of 10–12 individual barcoded libraries (100 pM each) were pooled and clonally amplified through emulsion PCR using the OneTouch Instrument and the Ion PGM Template OT2 200 Kit (Thermo Fisher Scientific). Emulsion breaking and enrichment were then performed. Finally, the sequencing of templates was performed after emulsion PCR on a PGM 318 chip using the Ion PGM 200 Sequencing Kit v2 according to the manufacturer’s instructions (200 bp read length; Thermo Fisher Scientific). Frozen and FFPE samples were run on separate chips, and the maximum number of samples on one chip was 12. Other methods are included Supplementary materials and methods.
Results
Somatic mutations among 50 cancer-related genes in OSCC cell lines
The CHPv2 panel targeted 207 amplicons covering genomic “hot spot” regions from 50 oncogenes and tumor suppressor genes that harbor 2855 COSMIC-cited mutations. Somatic mutation profiling of 18 OSCC cell lines was initially performed using this panel and semiconductor sequencing technology. The average number of reads per sample was 268,182 (range 112,123–554,643). The one-time average target coverage was 99.4%, with a raw mean accuracy of 99.1%. The average base coverage depth was 1129.9 (range 486–2289). Overall, we found 32 somatic mutations among six different genes: TP53 (17/18; 94.4%), CDKN2A (4/18; 22.2%), PIK3CA (3/18; 16.7%), SMAD4 (3/18; 16.7%), STK11 (1/18; 5.6%), and MET (1/18; 5.6%) (Figure 1, left). All 18 OSCC cell lines had one or more somatic mutations. In addition, a number of candidate driver genes were identified from the copy number variations (CNVs; Figure 1, right).
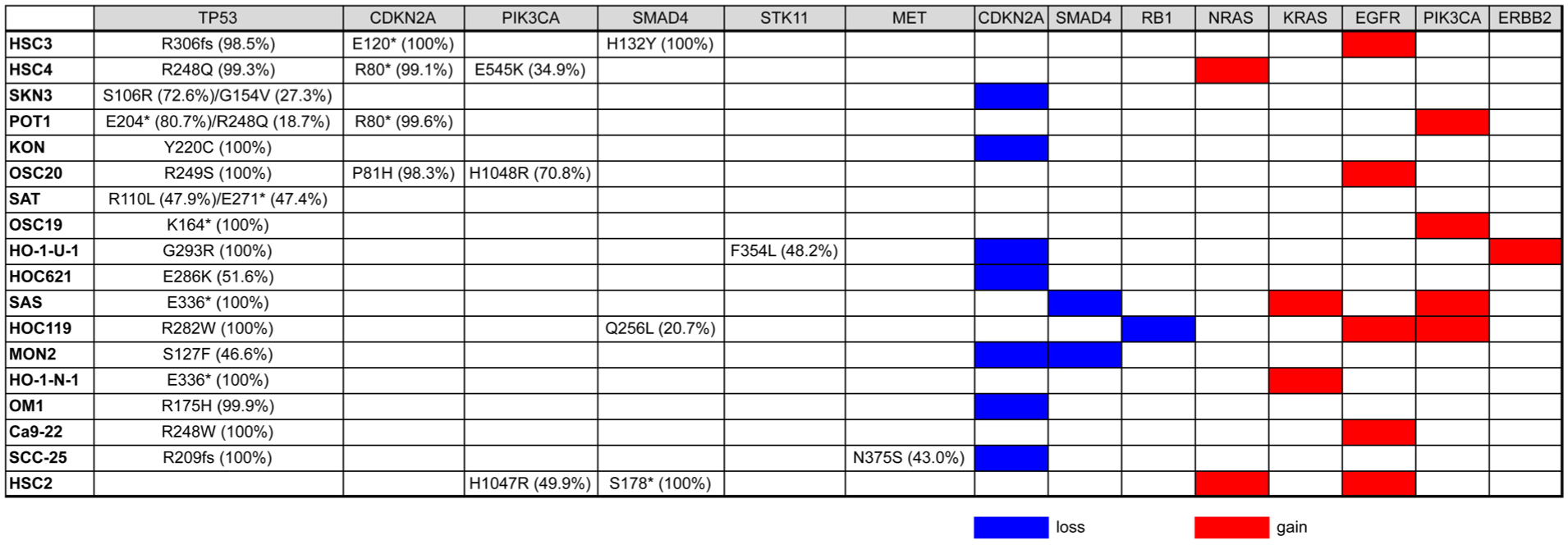
Mutations and CNVs found in 18 OSCC cell lines sequenced for 50 cancer-related gene hotspots. Mutations in cancer-associated genes were detected using Ion AmpliSeq Cancer Hotspot Panel v2. Percent mutant allelic frequencies are also listed. The CNVs were detected using the control sequence data provided by Thermo Fisher Scientific as a control, including both copy number gains and losses. Blanks indicate no alterations.
The analysis of ALK, RET, and ROS1 gene rearrangements has become an area of focus for targeted therapies in non-small cell lung cancers. 10 ALK rearrangements in lung squamous cell carcinoma are rare but do exist.11–13 To our knowledge, there have been no reports of analyses of these rearrangements in OSCC. We then performed high-throughput genotyping of 70 variants of ALK, RET, and ROS1 fusion genes (AmpliSeq RNA Lung Cancer Research Fusion Panel). As a result, we uncovered no fusion-positive OSCC cell lines (data not shown), suggesting that NSCLC-related gene fusions are rare in OSCC.
Quality check of FFPE samples
A total of 95 FFPE samples of primary OSCCs were used for DNA extraction and quantified via real-time PCR of a short amplicon (87 bp) for the RNase P gene. A total of 21 samples (22.1%) with low DNA concentrations (<0.85 ng/µL) were withdrawn, as recommended by the manufacturer, due to low DNA yield. We performed library preparation for 74 samples and then sequenced these libraries using CHPv2. Successful sequencing was characterized by the following sequencing metrics: on-target reads (>75%), coverage uniformity (>75%), and average coverage depth (>250×). According to the results, 36 samples failed among the 74 sequenced samples. Out of 95 FFPE samples, 57 (60%) were ineligible for NGS analyses.
We then retrospectively evaluated the factors that may affect the success or failure of sequencing using FFPE samples. Although DNA quantification is achieved by quantitative polymerase chain reaction (qPCR) using a short amplicon, poor DNA quality dramatically reduces the PCR amplification efficiency of long targets. Therefore, the ratio between the concentrations of the 256-bp long amplicon and the 87-bp short amplicon (long/short ratio) was useful as a measure of DNA quality. Although a significantly lower mean long/short ratio was obtained for the failed samples than for the successful samples (0.03 vs 0.20, p = 0.0002), several cases with long/short ratios of 0 were successfully sequenced (Figure 2(a)). The mean library concentration was 748.6 pM for the success group and 28.0 pM for the failure group (Figure 2(b)). Sequencing success correlated positively with the library concentration (p < 0.0001). We then validated the prepared amplicon libraries using the Agilent High Sensitivity DNA Kit and the Bioanalyzer 2100 instrument. As shown in Figure 2(c), all samples with a typical size distribution were successfully sequenced, suggesting that library concentration and size distribution may be useful guidelines for successful results. In contrast, four out of four samples with an unexpected broad peak failed the analysis. Interestingly, although the yield was very low for several samples, as shown in the lower panel of Figure 2(c), we were able to obtain viable libraries in sufficient quantity for sequencing.

Quantitation of FFPE samples for which sequencing was successful or failed. (a) The relative quality of FFPE samples was calculated by normalizing the concentration of the 256-bp amplicon to that of the 87-bp amplicon. The data are represented as a scattered dot plot with a line at the mean. (b) The library concentration was determined by real-time PCR. (c) The library size and range were visualized using a Bioanalyzer High Sensitivity DNA Kit.
Somatic mutations among 50 cancer-related genes in OSCC tissues
A total of 80 OSCC samples (38 FFPE and 42 frozen samples; Table 1) were analyzed. Of the 80 (40%) patients, 32 had human papillomavirus (HPV)-positive OSCC (Supplementary Table S1). A sequencing overview of the successfully sequenced tumors is shown in Supplementary Table S2. Mean coverage depths of 940.6 and 1493.4 reads per base were observed for FFPE samples (n = 38) and frozen samples (n = 42), respectively. Compared with the frozen samples, the FFPE samples had a decreased percentage of on-target reads and uniformity (Supplementary Tables S2 and S3).
Patient characteristics of OSCC (n = 80).

OSCC: oral squamous cell carcinoma; FFPE: formalin-fixed paraffin-embedded.
Among all cases, 76 mutations were detected in 15 genes (Figure 3(a) and Supplementary Figure S1). No mutations were detected in 24 (30.0%) of the OSCC samples. There was no correlation between the average coverage depth and the number of mutations (data not shown). TP53 was the most commonly mutated gene, with variants identified in 40 of 80 cases (50.0%). The next most commonly mutated genes were CDKN2A (16.3%), PIK3CA (7.5%), HRAS (5.0%), MET (2.5%), and STK11 (2.5%). Other variants detected in one case included hotspot mutations in ALK, FBXW7, FGFR2, NOTCH1, CDH1, PTEN, SMAD4, PDGFRA, and APC. No mutations were identified within the hotspot regions of the other 35 genes. A single mutation was identified in 39 cases (48.8%), two mutations in 14 cases (17.5%), and three mutations in 3 cases (3.8%) (Supplementary Figure S1).

Frequencies of somatic hotspot mutations in patients with OSCC: (a) the overall frequencies of gene mutations (n = 80) and (b) the associations of clinicopathological variables with TP53 mutation.
TP53 mutations were significantly correlated with the stage of disease and metastatic lymph nodes in OSCC patients (p < 0.05; Figure 3(b)). Of the TP53 mutations, 32 (80.0%) were predicted to be missense mutations, 5 (12.5%) nonsense mutations, and 3 (7.5%) frameshift mutations. In addition, the majority of TP53 mutations (38/40; 95.0%) were localized in the DNA binding domain of the protein (Figure 4).

Mutation distribution in the functional domains of TP53 (n = 40). The mutation distribution in the functional domains of TP53 is shown. Any position with a mutation contains a circle (green, missense mutation; black, nonsense and frameshift mutations), and the length of the line depends on the number of mutations detected at that codon. The colored boxes are specific functional domains. Above the lollipops, the frequent variants are annotated as the amino acid change at that specific site.
Mutations were more common in HPV-negative than in HPV-positive OSCCs with 1.15 versus 0.66 mutations/tumor, respectively, and were detected in 79.2% (38/48) of HPV-negative and 50.0% (16/32) of the HPV-positive cases (p = 0.0064; Table 2). This difference was mainly due to the difference in TP53 mutations. TP53 mutations tended to be more frequent in HPV-negative OSCCs compared to HPV-positive cases (32/48, 66.7% vs 8/32, 25.0%; p = 0.00026). In addition, mutant TP53 was found much less frequently in OSCC specimens that contained HPV16 DNA (16.7%; Supplementary Table 4).
Clinicopathological features according to HPV infection status.

HPV: human papillomavirus; N.S.: not significant.
CNVs in targeted genes
We also detected CNVs in the segments of the genome that could be duplicated or deleted from sequencing data. A number of candidate driver genes were identified from the CNVs (Figure 5). The most frequently gained gene was EGFR (37.5%), followed by NRAS (33.8%), PIK3CA (27.5%), FGFR3 (17.5%), KIT (12.5%), MET (11.3%), and ERBB2 (8.8%). In contrast, the most frequently lost gene was RB1 (28.8%), followed by PTEN (21.3%), CDKN2A (21.3%), APC (15.0%), SMAD4 (13.8%), ATM (11.3%), STK11 (6.3%), and TP53 (6.3%). We validated 86.4% (19/22) and 100% (7/7) of the OSCC tissues that showed CNV gain for PIK3CA and ERBB2, respectively, in quantitative PCR copy number analysis (Supplementary Figure S2). All variant calls and CNVs are available in Supplementary Figure S1.

Frequencies of CNVs in patients with OSCC. The CNVs were detected using the control sequence data provided by Thermo Fisher Scientific as a control, including both copy number gains and losses. The overall frequencies of copy number gains and losses are shown (n = 80).
Circulating cell-free DNA analysis
Circulating cell-free DNA (cfDNA) is a promising tool for use as a non-invasive biomarker for cancer mutation profiling.14,15 Blood samples for plasma cfDNA isolation were collected prospectively from two OSCC patients (Patient No. 10, stage III; Patient No. 27, stage IVa) before and after radical surgery. We performed a similar targeted NGS analysis of plasma DNA samples, with a mean coverage depth of over 10,000×. We detected the tumor-specific TP53 mutations R248Q (No. 10) and G266R (No. 27) in the plasma DNA before the patients underwent surgery. Interestingly, TP53 mutations from cfDNA became undetectable 2 weeks after surgery (Supplementary Figures S3 and S4). The patients currently remain alive, without recurrence, at 12 months after the surgery. This result demonstrated the feasibility of clinically useful targeted NGS-based circulating tumor DNA (ctDNA) mutation profiling to guide treatment decisions in cancer.
Discussion
The NGS platform can be applied to clinical diagnostic assays with different levels of complexity, including small hotspot panels, tumor-specific gene panels, WES, and possibly whole-genome and/or whole-transcriptome sequencing. In this study, a hotspot mutation analysis was performed in 80 Japanese patients with OSCC. Our approach using the Ion Torrent platform facilitates the simultaneous high-throughput detection of somatic mutations and CNVs in OSCC samples, including FFPE. Only small amounts (10 ng) of input DNA are required. Moreover, library preparation, sequencing, and data analysis can all be performed within 48 h, and this amplicon workflow is considerably faster than capture-based target sequencing. 16
We recently validated a 409-gene comprehensive panel for 47 OSCC patients. 17 The panel, the Ion AmpliSeq Comprehensive Cancer Panel (CCP), consists of approximately 16,000 primer pairs covering all exons of 409 genes with known cancer associations. In our previous paper, an average of 5.49 non-synonymous mutations (range: 0–12) were detected in OSCC by CCP, suggesting that CHPv2 does not cover all mutations in OSCC patients. Among the 50 genes included in the CHPv2, we found frequent mutations in the TP53 (61.7%), NOTCH1 (25.5%), CDKN2A (19.1%), and PIK3CA (10.6%) genes by CCP. 17 In this study, only one case out of 80 OSCCs (1.3%) exhibited a NOTCH1 mutation, which was inconsistent with the results of our previous study. The NOTCH1 coding sequence contains 34 exons, and the genomic sequence spans 48 kb. CCP covers all exons of the NOTCH1 gene, with a total of 91 amplicons (data not shown). The overall coverage of the NOTCH1 coding sequence in CCP was 94.6%. In contrast, CHPv2 includes only three amplicons, including exons 26, 27, and 34, suggesting the presence of mutations in an uncovered region of the NOTCH1 gene. We also noticed that CHPv2 only targets half of the TP53 coding regions (exons 2–10) covered by CCP (data not shown).
Chen et al. 18 sequenced 737 loci from 45 cancer-related genes in 345 advanced OSCC samples using the CHPv1 panel. The four most frequently mutated genes in our cohort were TP53, CDKN2A, PIK3CA, and HRAS from a group of 50 genes included in the CHPv2 panel, which is consistent with the findings of Chen et al. Because they used the cutoff values of more than 3% variant frequency, the prevalence of these mutated genes was higher than that detected in our study. Importantly, HPV-negative OSCCs were associated in our study with TP53 mutations. More recently, Oikawa et al. 19 reported the targeted sequencing of 220 OSCC tissues using the same 50-gene CHPv2 panel. The genetic mutations in that study exhibited similar frequencies of mutations in CDKN2A (15.9%) and PIK3CA (8.6%), whereas mutations in TP53 were less frequent (35.9%). The frequency of TP53 mutations in our samples was slightly lower than that observed in previous WES studies of OSCC (62%–73%),20,21 which may be due to the fact that the CHPv2 panel covers only half of the coding region of TP53 (data not shown).
Although NOTCH1 is one of the commonly mutated genes in squamous cell carcinoma, including HNSCC, with a mutation rate of 17.8% in The Cancer Genome Atlas (TCGA) cohort (http://www.cbioportal.org/study?id=hnsc_tcga#summary), only one case (1.3%) was found to be positive for NOTCH1 mutations in this study. Notch signaling has both oncogenic and tumor-suppressive roles, depending on the cellular context. 22 NOTCH1 plays the role of oncogene in liquid tumors, such as lymphomas and leukemias.23,24 In contrast, NOTCH1 is suggested to be a tumor suppressor gene in solid tumors, including OSCC,25–27 in which inactivating mutations may be distributed evenly throughout the gene. However, the CHPv2 panel only targeted NOTCH1 hotspot regions found in liquid tumors (three amplicons). Therefore, the failure to detect a NOTCH1 mutation may be explained by the fact that only a limited number of NOTCH1 exon regions were sequenced by the assay in this study. Moreover, the panel used in this study does not include some important genes that have been detected in WES studies of OSCC, such as CASP8, EPHA2, TP63, and FAT1.21,28 Thus, although many mutations in cancer-related genes can be detected using this panel, the clinical sensitivity may be reduced due to the restricted nature of the hotspot panel, indicating that the coverage of the full coding sequence is ideal for tumor suppressor genes.
Panel sequencing of routine pathology materials, such as FFPE samples, can yield information on several driver mutations, including some for which targeted therapies are available. In this study, all frozen samples were successfully sequenced. In contrast, the sequencing failure rate of FFPE samples was 51.4% (38/74). This failure may be attributed to formalin fixation–induced degradation, indicating the limitation of conventional methods when using FFPE samples. Because inadequate DNA quality has a great impact on the amplification efficiency of longer targets, 29 FFPE DNA was quantitated using amplicons of two different sizes (87 bp (short) and 256 bp (long)). As shown in Figure 2(a), the long/short ratio may be used to predict the success of targeted NGS sequencing. Surprisingly, 37.0% (20/54) of the samples with low long/short ratios (<0.1) were successfully analyzed. Moreover, the low library concentration effectively prevented the sequencing of poor-quality samples (Figure 2(b)). We noticed that the DNA Bioanalyzer assays could be used to predict the success of NGS library preparation (Figure 2(c)), suggesting that more than one validation should be used for quality management in NGS.
ctDNA extracted from blood has become a clinically feasible biomarker in various types of cancer. However, the clinical significance of ctDNA in OSCC among Japanese populations requires further investigation. In this study, using the same CHPv2 panel, we sequenced plasma cfDNA (which includes ctDNA, along with cfDNA from other sources) from two advanced OSCC patients before and after surgery (Supplementary Figures S3 and S4). These patients harbored the same TP53 mutations (R248Q in No. 10 and G266R in No. 27) in the primary tumor and pre-surgical plasma cfDNA. Importantly, TP53 mutations in the plasma of these patients were undetectable after surgery. The results suggested that the monitoring of mutations in ctDNA might be useful as a surrogate for imaging in order to evaluate treatment efficacy and may increase timely therapeutic reorientation, although cfDNA mutations should be validated using a highly sensitive digital PCR technique in future studies.
Finally, the amplicon sequence platform using the semiconductor sequencer is readily scalable so that specific gene panels may be designed for any cancer type. The speed and simplicity of this platform are well suited to a clinical setting. Coupling multiplex PCR-based library preparation with NGS therefore provides an important advance toward the implementation of routine clinical sample typing and precision medicine for cancer patients. The main limitation of the mutational analysis using a small hotspot panel is the absence of any relevant targetable mutations among the analyzed genes. In the near future, a customized gene panel covering hotspots or complete coding regions of genes that are more frequently mutated in OSCC may become a diagnostic tool to more precisely predict prognosis and better guide treatment decisions in the context of genetic-based risk assessment.
Supplemental Material
Fig._S1 – Supplemental material for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma
Supplemental material, Fig._S1 for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma by Takafumi Nakagaki, Miyuki Tamura, Kenta Kobashi, Akina Omori, Ryota Koyama, Masashi Idogawa, Kazuhiro Ogi, Hiroyoshi Hiratsuka, Takashi Tokino and Yasushi Sasaki in Tumor Biology
Supplemental Material
Fig._S2 – Supplemental material for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma
Supplemental material, Fig._S2 for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma by Takafumi Nakagaki, Miyuki Tamura, Kenta Kobashi, Akina Omori, Ryota Koyama, Masashi Idogawa, Kazuhiro Ogi, Hiroyoshi Hiratsuka, Takashi Tokino and Yasushi Sasaki in Tumor Biology
Supplemental Material
Fig._S3 – Supplemental material for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma
Supplemental material, Fig._S3 for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma by Takafumi Nakagaki, Miyuki Tamura, Kenta Kobashi, Akina Omori, Ryota Koyama, Masashi Idogawa, Kazuhiro Ogi, Hiroyoshi Hiratsuka, Takashi Tokino and Yasushi Sasaki in Tumor Biology
Supplemental Material
Fig._S4 – Supplemental material for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma
Supplemental material, Fig._S4 for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma by Takafumi Nakagaki, Miyuki Tamura, Kenta Kobashi, Akina Omori, Ryota Koyama, Masashi Idogawa, Kazuhiro Ogi, Hiroyoshi Hiratsuka, Takashi Tokino and Yasushi Sasaki in Tumor Biology
Supplemental Material
Supplementary_materials_and_methods-rerevise – Supplemental material for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma
Supplemental material, Supplementary_materials_and_methods-rerevise for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma by Takafumi Nakagaki, Miyuki Tamura, Kenta Kobashi, Akina Omori, Ryota Koyama, Masashi Idogawa, Kazuhiro Ogi, Hiroyoshi Hiratsuka, Takashi Tokino and Yasushi Sasaki in Tumor Biology
Supplemental Material
TUB-18-0041.R2_TableS1 – Supplemental material for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma
Supplemental material, TUB-18-0041.R2_TableS1 for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma by Takafumi Nakagaki, Miyuki Tamura, Kenta Kobashi, Akina Omori, Ryota Koyama, Masashi Idogawa, Kazuhiro Ogi, Hiroyoshi Hiratsuka, Takashi Tokino and Yasushi Sasaki in Tumor Biology
Supplemental Material
TUB-18-0041.R2_TableS2 – Supplemental material for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma
Supplemental material, TUB-18-0041.R2_TableS2 for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma by Takafumi Nakagaki, Miyuki Tamura, Kenta Kobashi, Akina Omori, Ryota Koyama, Masashi Idogawa, Kazuhiro Ogi, Hiroyoshi Hiratsuka, Takashi Tokino and Yasushi Sasaki in Tumor Biology
Supplemental Material
TUB-18-0041.R2_TableS3 – Supplemental material for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma
Supplemental material, TUB-18-0041.R2_TableS3 for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma by Takafumi Nakagaki, Miyuki Tamura, Kenta Kobashi, Akina Omori, Ryota Koyama, Masashi Idogawa, Kazuhiro Ogi, Hiroyoshi Hiratsuka, Takashi Tokino and Yasushi Sasaki in Tumor Biology
Supplemental Material
TUB-18-0041.R2_TableS4 – Supplemental material for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma
Supplemental material, TUB-18-0041.R2_TableS4 for Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma by Takafumi Nakagaki, Miyuki Tamura, Kenta Kobashi, Akina Omori, Ryota Koyama, Masashi Idogawa, Kazuhiro Ogi, Hiroyoshi Hiratsuka, Takashi Tokino and Yasushi Sasaki in Tumor Biology
Footnotes
Acknowledgements
Takafumi Nakagaki and Miyuki Tamura contributed equally to this work. We thank all the members and staff for their contributions to the sample collection and the completion of our study.
Author note
Yasushi Sasaki is now affiliated to Biology Division, Department of Liberal Arts and Sciences, Center for Medical Education, Sapporo Medical University, Sapporo, Japan.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
This study was approved by the Institutional Review Boards of Sapporo Medical University (reference no. 26-25 from the Sapporo Medical University Ethics Committee). All patients provided written informed consent. The study was conducted in accordance with the ethical principles of the Declaration of Helsinki.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by JSPS KAKENHI under Grant No. 25430115 and JSPS KAKENHI under Grant No. 16H06279.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.