
Abstract
Objectives:
Preoperative chemoradiation is currently the standard of care in locally advanced rectal carcinoma, even though a subset of rectal tumors does not achieve major clinically meaningful responses upon neoadjuvant chemoradiation. At present, no molecular biomarkers are available to predict response to neoadjuvant chemoradiation and select resistant tumors willing more intense therapeutic strategies. Thus, BRAF mutational status was investigated for its role in favoring resistance to radiation in colorectal carcinoma cell lines and cyclin-dependent kinase 1 as a target to improve radiosensitivity in BRAF V600E colorectal tumor cells.
Methods:
Colony-forming assay and apoptotic rates were evaluated to compare the sensitivity of different colon carcinoma cell lines to ionizing radiation and their radiosensitivity upon exposure to BRAF and/or cyclin-dependent kinase 1 inhibitory/silencing strategies. Cyclin-dependent kinase 1 expression/subcellular distribution was studied by immunoblot analysis.
Results:
Colon carcinoma BRAF V600E HT29 cells exhibited poor response to radiation compared to BRAF wild-type COLO320 and HCT116 cells. Interestingly, neither radiosensitizing doses of 5-fluoruracil nor BRAF inhibition/silencing significantly improved radiosensitivity in HT29 cells. Of note, poor response to radiation correlated with upregulation/relocation of cyclin-dependent kinase 1 in mitochondria. Consistently, cyclin-dependent kinase 1 inhibition/silencing as well as its targeting, through inhibition of HSP90 quality control pathway, significantly inhibited the clonogenic ability and increased apoptotic rates in HT29 cells upon exposure to radiation.
Conclusion:
These data suggest that BRAF V600E colorectal carcinoma cells are poorly responsive to radiation, and cyclin-dependent kinase 1 represents a target to improve radiosensitivity in BRAF V600E colorectal tumor cells.
Introduction
Resistance to ionizing radiation is one of the major obstacles for successful killing of cancer cells with radiotherapy. Indeed, radiation induces the activation of multiple signaling pathways, causing cancer cells to become inactivated and resulting in diverse types of stress responses, including apoptosis, cell cycle arrest, and senescence. 1 However, a subset of human malignancies fails to respond to radiotherapy, as they are resistant to radiation-induced apoptosis.1,2 Thus, the identification of novel mechanisms of resistance to radiation may aid to overcome radioresistance, improve radiotherapy efficacy and, ultimately, personalize treatments. 2
Preoperative chemoradiation is currently the standard of care for locally advanced rectal adenocarcinoma, obtaining tumor downstaging and lower rates of local failure. 3 Indeed, total mesorectal excision is curative for small rectal tumors, but the risk of locoregional recurrence, distant metastasis, and death increases with tumors extending through the muscularis propria (tumor-nodes-metastasis (TNM), T3 or T4) or with nodal involvement (TNM, N1 or N2). 3 In such a context, neoadjuvant chemoradiotherapy results in a wide spectrum of clinical responses and the magnitude of benefit is heterogeneous: while the achievement of pathological complete response is feasible in many patients, a subset of rectal carcinomas are not fully responsive to radiation. 4 Thus, the identification of biomarkers predictive of resistance to preoperative chemoradiation and the characterization of novel targets to improve response to radiotherapy are area of active research in rectal cancer.
Advanced colorectal carcinomas (CRCs) bearing the BRAF V600E mutation are largely acknowledged as aggressive malignancies with poor prognosis and lack of response to anticancer therapies. 5 By contrast, conflicting results have been proposed on the role of BRAF mutations in driving the sensitivity of rectal carcinoma cells to radiation, with some studies showing poor responses and others no influence.6–9 Thus, this study was designed to evaluate in vitro the radiosensitivity of CRC cells with different RAS/BRAF mutational profiles and exploit cyclin-dependent kinase 1 (CDK1) targeting as a strategy to improve efficacy of radiation therapy in BRAF V600E tumor cells.
Materials and methods
Cell lines, chemicals, constructs, and siRNAs
Human CRC RAS/BRAF wild-type COLO320, KRAS G13D HCT116, and BRAF V600E HT29 cells were purchased from the American Type Culture Collection (ATCC LGS Standards, Sesto San Giovanni, Milan, Italy). Cell line authentication was verified by short tandem repeat (STR) profiling, according to ATCC product description. HCT116 and HT29 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1.5 mM glutamine, and 100 U/mL penicillin and streptomycin and COLO320 cells in RPMI 1640 supplemented with 10% FBS, 0.75 mM glutamine, and 100 U/mL penicillin and streptomycin.
Unless otherwise specified, reagents were purchased from Sigma-Aldrich (Milan, Italy). The BRAF inhibitor PLX4720 was purchased from Selleck Chemicals (Huston, USA); the HSP90/TNF receptor associated protein 1 (TRAP1) inhibitor, HSP990 was kindly provided by Novartis (Origgio, VA, Italy).
Wild-type BRAF and BRAF V600E constructs were kindly provided by Prof. Massimo Santoro (University of Naples Federico II, Naples, Italy). All the constructs were cloned in pcDNA3.1 vector (Invitrogen, Monza MB, Italy). Transient transfection of DNA plasmids was performed with PolyFect Transfection Reagent (Qiagen, Milan, Italy), according to the manufacturer’s protocol.
Small interfering RNAs (siRNAs) of BRAF and CDK1 were purchased from Qiagen (Cat. No. SI00299488, target sequence AACATATAGAGGCCCTATTGG, for BRAF; Cat. No. SI02663381 target sequence CAGGTTATATCTCATCTTTGA, for CDK1). For control experiments, cells were transfected with a similar amount of control siRNA (Qiagen; Cat. No. SI03650318, target sequence Qiagen properties). For knockdown experiments, siRNAs were diluted to a final concentration of 40 nM and transiently transfected by the HiPerFect Transfection Reagent (Qiagen), according to manufacturer protocol.
Cell culture irradiation
Cell cultures were irradiated using the Elekta Synergy Linear Accelerator; 6 MV photon energy was used with a 400-monitor unit (MU)/min dose rate. Because the maximum aperture of the linac field is 40 × 40 cm at source-surface distance (SSD) of 100, at most six plates were irradiated at a time. A phantom was constructed to minimize build-up effect and therefore improve scatter conditions in the medium and allow isodose coverage of 95–107%. The phantom was made of Plexiglas plates due to its tissue equivalent characteristics. The size of the phantom (40 × 40 × 1 cm) allowed sufficient scatter material around the radiation field to cover all plates; it was placed under the plates to allow a uniform posterior-anterior irradiation, avoiding the presence of air between the top of plates and cell cultures. Before starting our experiments, cell plates were placed on the phantom and were computed tomography (CT) scanned. CT data were imported into a treatment planning system (TPS), contoured, and planned with Oncentra Masterplan TPS (Elekta, Stockholm, Sweden). Spatial coordinates of set-up were established from CT data, and optimal geometry of the field was identified with TPS. An SSD of 100 cm and a gantry rotation of 180° and the interposition of the Plexiglas bolus were set. This arrangement was necessary to remedy the build-up phenomenon. Control samples were carried to the linac bunker but not irradiated in order to be exposed to the same conditions of transport and temperature.
MTT assays
Dimethylthiazol diphenyltetrazolium bromide (MTT; Sigma-Aldrich) dye assay was used as an in vitro cell viability test to evaluate sensitivity to radiation. 10 Briefly, cell lines were seeded into 24-well plates (2 × 104 cells/well), exposed to radiation as previously described and further incubated in standard medium for 24 h. Then, 600-µM MTT solution was added to each well and MTT incorporation was measured 3 h later using a BioTek microplate reader (model EL-340; BioMetallics, Princeton, NJ, USA).
Apoptosis assay
Apoptosis was evaluated by cytofluorimetric analysis of Annexin-V and 7-amino-actinomycin-D (7-AAD)-positive cells using the fluorescein isothiocyanate (FITC)-Annexin-V/7-AAD kit (Beckman Coulter, Milan, Italy). Stained cells were analyzed using the FACSCaliburTM (Becton Dickinson, Milan, Italy). Positive staining for Annexin-V as well as double staining for Annexin-V and 7-AAD was interpreted as signs of early and late phases of apoptosis, respectively. 11
Focus forming assays
Cells were seeded at a density of 300 cells/well in 6-well plates, 24 h later treated with specified pharmacological agents and/or exposed to radiation as reported in figure legends and left growing for 15 days with medium changes every 3 days. After 15 days from treatment, plates were fixed with methanol/acetic acid solution (1:7) and colored with crystal violet. Density of transformation foci were compared by cell counts and represented as average ± standard deviation (SD). 12
Immunoblot analysis
Total cell lysates were obtained by homogenization of cell pellets in a cold lysis buffer (20 mM Tris pH 7.5 containing 300 mM sucrose, 60 mM KCl, 15 mM NaCl, 5% (v/v) glycerol, 2 mM ethylenediaminetetraacetic acid (EDTA), 1% (v/v) Triton X-100, 1 mM PMSF, 2 mg/mL aprotinin, 2 mg/mL leupetin, and 0.2 % (w/v) deoxycholate) for 2 min at 4°C and further sonication for 30 s on ice. Mitochondrial and cytosolic fractions were purified by the Qproteome Mitochondria Isolation kit (Qiagen; Cat. No. 37612), according to the manufacturer protocol. Samples were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred on nitrocellulose membrane (Bio-Rad Laboratories GmbH, Munchen, Germany). Specific proteins were detected by using the following antibodies: mouse monoclonal anti-CDK1 (Santa Cruz Biotechnology, Heidelberg, Germany; sc-53219), rabbit polyclonal anti-pCDK1 (phosphoThr161, ab208915, Abcam, Cambridge, UK), mouse monoclonal anti-BRAF (Santa Cruz Biotechnology; sc-5284), mouse monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Santa Cruz Biotechnology; sc-69778), mouse monoclonal anti-phospho44/42 MAPK (pERK1/2, Cell Signaling Technology, ZA Leiden, Holland; #9106), rabbit polyclonal anti-MAPK 1/2 (ERK1/2, CalBiochem, #ABS44), and rabbit polyclonal anti-VDAC (Merck Millipore, #AB10527). Specific bands were revealed using the Clarity Western ECL Substrate (Bio-Rad Laboratories GmbH). Where indicated, protein levels were quantified by densitometric analysis using the Quantity One 4.5 software (Bio-Rad Laboratories GmbH). Band intensities were normalized with respect to each loading control and reported in Figures.
Statistical analysis
All experiments were independently performed at least three times and data reported as average ± SD. Two-sided Student’s t-test was used to establish the statistical significance between different levels of cell death. One-way analysis of variance (ANOVA) was applied to test the differences in cell survival among different groups. Because of multiple testing, Bonferroni correction was always applied. A probability value less than 0.05 (p < 0.05) was regarded as statistically significant. R statistical program was used for data analysis.
Results
BRAF V600E CRC cells are poorly sensitive to radiation
In preliminary experiments, the sensitivity of CRC cells to radiation was assessed in HCT116 cells exposed to increasing doses of radiation and evaluated for cell viability by MTT assay (Figure 1(a)). A dose-dependent inhibition of MTT incorporation was observed in irradiated compared to non-irradiated cells (Figure 1(a)). Since a statistically significant difference was already observed between control and 2 Gy radiation (p = 0.045), subsequent experiments were conducted using 2 Gy as minimal starting dose.

Colorectal carcinoma BRAF V600E HT29 cells are poorly responsive to radiation: (a) HCT116 cells were exposed to 2, 3, 4, and 8 Gy radiation and evaluated for cell viability by MTT assay. Statistical significance with respect to control cells: *p = 0.045; **p = 0.004; °p = 0.001; °°p = 0.0006. COLO320, HCT116, and HT29 cells were exposed to increasing doses of radiation (2–8 Gy) and evaluated for (b) clonogenic potential and (c) apoptotic cell death. Statistical significance with respect to HT29 cells exposed to the same experimental conditions: *p = 0.0002; °p = 0.039; **p = 0.0003; °°p = 0.0012. (d) Apoptotic cell death in COLO320 cells transfected with pMock, BRAF cDNA, or BRAF V600E mutant and exposed to 4 and 8 Gy radiation. Insert: BRAF immunoblot analysis in COLO320 cells transfected with pMock (1), BRAF cDNA (2), or BRAF V600E mutant (3). Statistical significance with respect to pMock transfected cells exposed to the same radiation dose: *p = 0.017; **p = 0.002.
Since BRAF V600E CRC cells are characterized by resistance to apoptosis and poor response to cytotoxic agents, 13 in further experiments RAS/BRAF wild-type COLO320, KRAS G13D HCT116 and BRAF V600E HT29 cells were exposed to increasing doses of radiation and evaluated for clonogenic potential (Figure 1(b)) and apoptotic cell death (Figure 1(c)). A dose-dependent inhibition of colony formation was observed in all cell lines upon exposure to radiation, being BRAF V600E HT29 cells characterized by more significant preservation of the clonogenic potential (Figure 1(b)). Indeed, a 2 Gy single dose reduced the surviving fraction of COLO320, HCT116, and HT29 cells to 35.7, 70.8, and 79.4% (p < 0.01, ANOVA test), respectively, and 4 and 8 Gy single doses showed a statistically significant difference in the clonogenic ability between HT29 and HCT116 and COLO320 cells (p < 0.01, ANOVA test; Figure 1(b)). In parallel experiments, the exposure to 4 and 8 Gy radiation induced significantly lower levels of apoptosis in HT29 cells than in COLO320 and HCT116 cells (p < 0.01, ANOVA test; Figure 1(c)) and in COLO320 cells transfected with BRAF cDNA or the BRAF V600E mutant compared to COLO320 cells transfected with empty vector (p < 0.01, ANOVA; Figure 1(d)). These data suggest that BRAF V600E HT29 cells are less sensitive to radiation than BRAF wild-type CRC cell lines.
Radiosensitization with 5-fluorouracil does not revert resistance of BRAF V600E CRC cells to radiation
Fluoropyrimidines are a well-known radiosensitizers, widely used in neoadjuvant protocols of rectal cancer chemoradiation. 14 Thus, the hypothesis that 5-fluorouracil (5FU) may revert resistance to radiation in BRAF V600E HT29 cells was tested. COLO320, HCT116, and HT29 cell lines were pretreated with increasing concentrations of 5FU (5–500 nM) for 15 h, exposed to 2 and 4 Gy single-dose radiation in the presence of 5FU and subsequently evaluated for their clonogenic potential. Interestingly, while 5FU produced a dose-dependent radiosensitizing effect in all cell lines, HT29 cells still showed lower sensitivity to radiation compared to BRAF wild-type cell lines, regardless the pretreatment with 5FU (Figure 2(a)–(c)). Interestingly, while the clonogenic potential of irradiated COLO320 and HCT116 cells was abolished by a pretreatment with 5FU in a concentration range between 15 and 500 nM 5FU, colony formation was still observed in HT29 cells treated with 500 nM 5FU before radiation (p < 0.05, for all comparisons among cell lines at each dose level, ANOVA test).

5-fluorouracil does not prevent resistance of colorectal carcinoma BRAF V600E HT29 cells to radiation: clonogenic potential of (a) COLO320, (b) HCT116, and (c) HT29 cells treated with increasing concentrations of 5FU (5–500 nM) for 15 h and subsequently exposed to 2 and 4 Gy radiations. Statistical significance with respect to HT29 cells exposed to the same experimental conditions: *p = 0.001; **p = 0.0012; °p = 0.0024; °°p = 0.0036; ***p = 0.006; °°°p = 0.012; ns, not significant.
BRAF targeting minimally induces radiosensitization in BRAF V600E colon carcinoma cells
To address the relevance of BRAF pathway in determining resistance of CRC cells to radiation, BRAF signaling was inhibited with the BRAF inhibitor PLX4720 (1 mM for 15 h) before irradiation. Of note, while PLX4720 inhibited BRAF downstream signaling as demonstrated by the suppression of ERK phosphorylation (Figure 3(a)), the colony-forming assay showed no statistical difference in the clonogenic potential of HT29 cells pretreated with PLX4720 and subsequently exposed to 4 or 8 Gy radiation compared to not-pretreated cells (Figure 3(c)). In parallel experiments, BRAF was silenced by siRNA in HT29 cells before irradiation (Figure 3(b)), and this resulted in a minimal sensitization of colon carcinoma cells to irradiation (Figure 3(d)).

BRAF targeting is ineffective in sensitizing colorectal carcinoma V600E HT29 cells to radiation. (a) ERK1/2 and pERK1/2 immunoblot analysis and (b) BRAF immunoblot analysis. (c) clonogenic potential in HT29 cells treated with 1 µM PLX4720 for 15 h and subsequently exposed to 4 and 8 Gy radiation and (d) clonogenic potential in HT29 cells transfected with control (siNeg) or BRAF siRNA (siBRAF) and subsequently exposed to 4 and 8 Gy radiation. Statistical significance respect to siNeg cells: *p = 0.01, **p = 0.004.
CDK1 targeting results in sensitization of BRAF V600E colon carcinoma cells to radiation
Since it has been widely suggested that mitochondrial CDK1 protects tumor cells from radiation-induced damages, 15 CDK1 expression and its subcellular distribution were evaluated in HT29 cells exposed to 1.5 Gy radiation. Of note, immunoblot analysis showed a slight increase of CDK1 protein levels in HT29 cells exposed to radiation and subsequently cultured in standard medium for 48 h (Figure 4(a)) and a more significant upregulation of its expression levels and its Thr161 phosphorylation in mitochondrial fractions of HT29 cells exposed to the same experimental conditions (Figure 4(b)). Interestingly, no upregulation of CDK1 was observed in BRAF wild-type HCT116 cells exposed to the same radiation dose and cultured up to 72 h after irradiation (Supplementary Figure 1). In order to establish whether CDK1 is involved in favoring resistance to radiation in BRAF-mutated CRC cells, HT29 cells were pretreated with the CDK1 inhibitor, RO3306, and evaluated for apoptotic rates (Figure 4(c)) and clonogenic potential (Figure 4(d)) upon radiation. Of note, while RO3306 did not induce apoptosis and minimally inhibited colony growth in non-irradiated cells, CDK1 inhibition significantly improved sensitivity to radiation. Indeed, RO3306 increased levels of apoptosis (Figure 4(c)) and reduced clonogenic potential (Figure 4(d)) in irradiated HT29 cells with high statistical significance. Consistently, CDK1 silencing enhanced radiation-induced apoptosis in HT29 cells (Figure 4(e)). Finally, since both CDK1 and BRAF are client proteins of HSP90 molecular chaperones,16,17 their stability was targeted using the dual HSP90/TRAP1 inhibitor, HSP990. 16 Thus, HT29 cells were pretreated with 75 or 150 nM HSP990 for 15 h and further exposed to radiation (Figure 5(a) and (b)). Indeed, HSP990 induced either a significant downregulation of CDK1 and BRAF expression (Figure 5(a)) or a parallel inhibition of the clonogenic potential in HT29 cells (Figure 5(b)). It is important to note that the pretreatment with HSP990 allowed the complete eradication of tumor cells upon exposure to 4 Gy radiation, as observed in COLO320 and HCT116 cells pretreated with 5FU (compare Figures 2(a)–(c) and 5(b)). No statistically significant difference was observed between different doses of HSP990 (75 versus 150 nM). These data suggest that CDK1 targeting represents a strategy to improve radiosensitivity in BRAF V600E colon carcinoma cells.

CDK1 targeting sensitizes colorectal carcinoma V600E HT29 cells to radiation: (a) CDK1 expression and (b) subcellular distribution in HT29 cells exposed to 1.5 Gy radiation and subsequently cultured in standard medium for 24–48 h (48 h in panel (b)). (c) Apoptotic rates and (d) clonogenic potential in HT29 cells pretreated with 2.5 or 5 µM RO3306 for 15 h (2.5 µM in panel (d)) and subsequently exposed to 2 and 4 Gy radiation. Statistical significance with respect to control cells exposed to radiation without pretreatment with RO3306: *p < 0.0001, **p = 0.001, °p = 0.04. (e) Apoptotic rates in HT29 cells transfected with control (siNeg) or CDK1 siRNA (siCDK1) and subsequently exposed to 2 Gy radiation. Statistical significance with respect to CDK1-silenced cells not exposed to radiation: *p < 0.0025. Insert: CDK1 immunoblot analysis in HT29 cells transfected with control (siNeg) or CDK1 siRNA (siCDK1). IR: ionizing radiation.
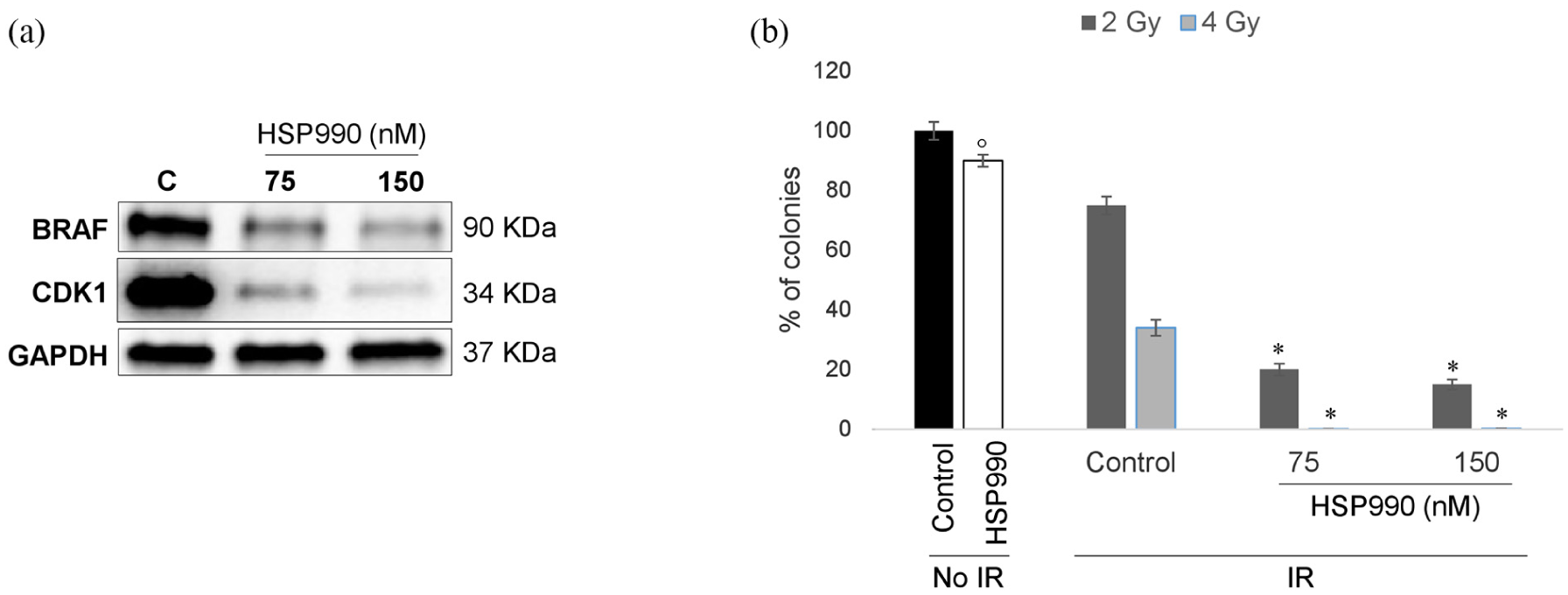
Targeting CDK1 and BRAF quality control sensitizes colon carcinoma V600E HT29 cells to radiation: (a) CDK1 and BRAF immunoblot analysis and (b) colony formation in HT29 cells pretreated with 75 or 150 nM HSP990 for 15 h and subsequently exposed to 2 and 4 Gy radiation. Control non-irradiated HT29 cells were treated with 150 nM HSP990 for 15 h. Statistical significance with respect to control cells not exposed to HSP990: *p = 0.0002; °p = 0.009. IR, ionizing radiation.
Discussion
Rectal carcinomas bearing BRAF mutations represent 2%–6% of total rectal epithelial tumors18–22 and are characterized by aggressive behavior and poor survival. 23 However, the impact of BRAF mutational status on response to preoperative chemoradiation is still controversial, with some studies showing low pathological complete response rates in RAS- or BRAF-mutated tumors6,7 and others no impact8,9 and this is likely due to the small number of BRAF-mutated rectal carcinomas in these clinical series. In such a context, the question whether BRAF oncogenic mutations enhance the apoptotic threshold of rectal cancer cells and impact on response to radiation is extremely relevant, since it is established that human BRAF V600E CRCs are resistant to apoptosis and poorly responsive to standard chemotherapeutics and molecular targeted agents. 13 Thus, this study was designed to evaluate the sensitivity of BRAF V600E CRC cells to radiation and identify molecular mechanisms of radioresistance. Our data suggest that: (a) BRAF V600E HT29 CRC cells are poorly sensitive to radiation and chemoradiation, (b) BRAF targeting is ineffective in restoring sensitivity to radiation, and (c) CDK1 may represent a target to improve radiosensitivity in BRAF-mutated CRC cells.
Radioresistance is a serious concern in daily clinical practice, causing radiotherapy failure and subsequent tumor relapse. Indeed, several human malignancies fail to respond to radiotherapy or chemoradiotherapy due to activation of survival pathways and/or selection of tumor subclones with intrinsic capacity to resist to apoptosis.24,25 Thus, the identification of mechanisms of poor response to radiation would allow the design of novel radiosensitizing strategies to overcome tumor radioresistance and thus improve the outcome of radiotherapy. In such a context, our data suggest that BRAF V600E colorectal tumor cells are poorly sensitive to ionizing radiation compared to BRAF wild-type cell lines. Indeed, the constitutive activation of RAS/RAF/ERK axis is responsible for survival responses in cancer cells, thus enhancing the apoptotic threshold and favoring resistance to chemotherapeutics.26,27 Consistently with this view, we recently reported a bidirectional crosstalk between BRAF signaling and the mitochondrial molecular chaperone TRAP1,12,16 involved in resistance to apoptosis and chemotherapeutics in human CRCs.28–30 Indeed, BRAF quality control is regulated by TRAP1 network, and its signaling is enhanced in human CRCs with high TRAP1 expression. 16 In addition, TRAP1 is tyrosine phosphorylated upon activation of BRAF signaling, which results in increased antiapoptotic responses. 12 Thus, while our data support the hypothesis that BRAF V600E mutations favor poor response to ionizing radiation in vitro, further studies are needed to establish whether BRAF mutational status represents a predictive tool to select human rectal carcinomas poorly responsive to preoperative chemoradiation and thus suitable for more aggressive treatments.
Consistently with the current literature, BRAF targeting did not show radiosensitizing activity in colorectal tumor cells. Indeed, it is established that BRAF inhibitors are poorly active in BRAF-mutated CRCs and only the combined inhibition of BRAF and other signaling molecules (i.e., EGFR or MEK) produces clinically meaningful responses in human CRCs bearing BRAF mutations. 31 Thus, in the attempt to identify novel targets to improve radiation activity in BRAF-mutated tumor cells, we observed that CDK1 is likely involved in resistance to radiation, being CDK1 upregulated/relocated in mitochondria of HT29 cells exposed to radiation. Our observation is consistent with previous evidence showing that the key cell cycle regulators, including cyclin D1/CDK4 and cyclin B1/CDK1 complexes, are responsible for regulation of mitochondrial antiapoptotic pathway and, thus, favor radioresistance. 15 Mechanistically, cyclin B1/CDK1 complex induces resistance to ionizing radiation, enhancing mitochondrial bioenergetics to meet the increased cellular fuel demand for DNA repair and cell survival under genotoxic stress. 15 Indeed, upon radiation, CDK1 relocates to mitochondria and boosts adenosine triphosphate (ATP) generation, thus favoring DNA repair and cell survival. 32 In addition, cyclin B1/CDK1 regulates manganese superoxide dismutase (MnSOD) through its Ser106 phosphorylation, and this results in increased antioxidant responses, improved mitochondrial function, and resistance to radiation-induced apoptosis. 33 Crucial in CDK1-dependent adaptive radioresistance is SIRT3 transcription and post-translational modifications, since SIRT3 enzymatic activity is enhanced via Thr150/Ser159 phosphorylation by cyclin B1/CDK1, which is also induced by radiation and relocates to mitochondria together with SIRT3. 34 Consistently, our data suggest that the level of the phosphorylated active form of CDK1 is increased in mitochondria upon radiation and that the CDK1-specific inhibitor, RO3306 or CDK1 silencing improves radiosensitivity in BRAF V600E CRC cells. This observation is also consistent with the radiosensitizing activity showed by AZD5438, a highly specific inhibitor of CDK1, 2, and 9, in radioresistant non–small cell lung carcinoma cell lines. 35 Finally, CDK1 and BRAF quality control pathway targeted by the dual HSP90/TRAP1 inhibitor, HSP990 16 resulted in even stronger radiosensitizing activity, supporting the conclusion that HSP90 molecular chaperone inhibitors are potential radiosensitizing agents in BRAF-mutated rectal cancer cells, as previously observed in glioblastoma cells. 36 It is important to note that only the exposure to HSP990 allowed the complete eradication of BRAF V600E HT29 cells upon radiation, as observed in BRAF wild-type CRC cells treated with 5FU as radiosensitizer. However, it should be also emphasized that our data do not allow the conclusion that this mechanism of radioresistance is specific for BRAF-mutated colorectal tumor cells, even though it is intriguing that KRAS-mutated HCT116 cells did not show any modification of CDK1 level in response to radiation. In such a perspective, significant differences are emerging in terms of signal transduction networks and their impact on treatment outcome between oncogenic KRAS and BRAF in CRC cells, 37 even though the molecular mechanism responsible for the differential expression/phosphorylation of CDK1 in mitochondria of KRAS- and BRAF-mutated cell lines is still unclear and an object of further investigation.
In conclusion, this study provides the proof of concept that BRAF V600E CRC cells are poorly sensitive to ionizing radiation and that targeting CDK1 pathway may represent a strategy to improve radiosensitivity in this tumor-cell model. Further studies are needed to establish whether CDK1 or HSP90 chaperone targeting may provide a strategy to personalize/improve neoadjuvant chemoradiation in human rectal cancers.
Supplemental Material
Supplementary_figure_1 – Supplemental material for Cyclin-dependent kinase 1 targeting improves sensitivity to radiation in BRAF V600E colorectal carcinoma cells
Supplemental material, Supplementary_figure_1 for Cyclin-dependent kinase 1 targeting improves sensitivity to radiation in BRAF V600E colorectal carcinoma cells by Girolamo Spagnoletti, Valeria Li Bergolis, Annamaria Piscazzi, Francesca Giannelli, Valentina Condelli, Lorenza Sisinni, Giuseppe Bove, Giovanni Storto and Matteo Landriscina in Tumor Biology
Footnotes
Acknowledgements
Girolamo Spagnoletti, Matteo Landriscina, and Giuseppe Bove contributed to study concept and design. Girolamo Spagnoletti, Valeria Li Bergolis, Annamaria Piscazzi, Francesca Giannelli, and Valentina Condelli contributed to data acquisition. Girolamo Spagnoletti, Valeria Li Bergolis, Annamaria Piscazzi, Valentina Condelli, Lorenza Sisinni, Giovanni Storto, and Matteo Landriscina contributed to the analysis and interpretation of data. Girolamo Spagnoletti and Matteo Landriscina contributed in drafting of the manuscript. Girolamo Spagnoletti, Valeria Li Bergolis, Annamaria Piscazzi, Valentina Condelli, Lorenza Sisinni, Giovanni Storto, and Matteo Landriscina gave the critical revision of the manuscript., Matteo Landriscina obtained funding and was responsible for study supervision.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by AIRC Grant IG2015 Id.16738 to Matteo Landriscina.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.