
Abstract
Recurrence of breast cancer after radiotherapy may be partly explained by the presence of radioresistant cells. Thus, it would be desirable to develop an effective therapy against radioresistant cells. In this study, we demonstrated the intense antitumor activity of cytokine-induced killer cells against MCF-7 and radioresistant MCF-7 cells, as revealed by cytokine-induced killer–mediated cytotoxicity, tumor cell proliferation, and tumor invasion. Radioresistant MCF-7 cells were more susceptible to cytokine-induced killer cell killing. The stronger cytotoxicity of cytokine-induced killer cells against radioresistant MCF-7 cells was dependent on the expression of major histocompatibility complex class I polypeptide–related sequence A/B on radioresistant MCF-7 cells after exposure of cytokine-induced killer cells to sensitized targets. In addition, we demonstrated that cytokine-induced killer cell treatment sensitized breast cancer cells to chemotherapy via the downregulation of TK1, TYMS, and MDR1. These results indicate that cytokine-induced killer cell treatment in combination with radiotherapy and/or chemotherapy may induce synergistic antitumor activities and represent a novel strategy for breast cancer.
Introduction
Radiotherapy has a well-established role in the treatment of patients with locally advanced breast cancer. 1 However, breast cancer cells are noted to undergo the development of radioresistance and enrichment of breast cancer stem cell subpopulation following radiation therapy. 2 Thus, targeting radioresistant breast cancer cells is associated with a significant reduction in tumor recurrence and improved overall survival.
Adoptive immunotherapies have been tested for the treatment of various tumors that may be refractory to conventional therapies. 3 Adoptive immunotherapy approaches with cytokine-induced killer (CIK) cells are particularly promising for successful application in cancer treatment.4,5 CIK cells are generated by culturing peripheral blood mononuclear cells (PBMCs) with the addition of interferon gamma (IFN-γ), anti-CD3 antibody, and interleukin-2 (IL-2).6,7 The obtained CIK cell population is mainly a CD3+ heterogeneous T-cell population where it is possible to distinguish two predominant subsets, respectively, positive (CD3+CD56+) and negative (CD3+CD56−) for the membrane expression of CD56. CD3+CD56+ cells represent T-cell subset with the greatest cytotoxicity in the CIK cell population.8,9 Advantages of these cells are their high cytotoxicity even at low cell numbers, their high proliferative rate, their non–major histocompatibility complex (non-MHC)–restricted cytotoxicity, and their activity against multidrug resistant tumor cell lines.10,11
In this study, we have assessed whether radioresistant breast cancer cells are sensitive to CIK cell killing. We demonstrate an enhanced ability of CIK cells to kill radioresistant MCF-7 (MCF-7/R) cells, to inhibit tumor cell proliferation and invasion compared with MCF-7 cells. We further elucidate that the mechanisms responsible for killing MCF-7/R cells more effectively involve the overexpression of MHC class I polypeptide–related sequence A/B (MICA/B) on tumor cells. In addition, we showed that CIK cell treatment could sensitize breast cancer cells to chemotherapy.
Materials and methods
Cell culture
MCF-7/R cells were generated from MCF-7 cells as previously described. 12 MCF-7 cells were exposed to fractionized ionizing irradiation with a total dose of 60 Gy (2 Gy per fraction, five times per week for 6 weeks). MCF-7 and MCF-7/R cells were cultured in Eagle’s minimum essential medium (EMEM) that was supplemented with 10% fetal bovine serum (FBS), 5% sodium pyruvate, 5% nonessential amino acid, penicillin (100 U/mL), and streptomycin (100 mg/mL) in a 37°C incubator.
CIK cell culture
Human peripheral blood samples were obtained from healthy volunteer blood donors. All individuals provided their informed consent. PBMCs were isolated on Ficoll Hypaque (GE Healthcare Life Sciences, Shanghai, China). PBMCs were incubated in RPMI-1640 medium supplemented with 20 ng/mL of anti-CD3 antibody (R&D Systems, Minneapolis, MN, USA), 100 U/mL of rhIL-2 (Jinan Quanqi, Jinan, China), and 200 U/mL of IFN-γ (Shanghai Kaimao, Shanghai, China). The medium was replaced with fresh IL-2 and IFN-γ-containing medium every 5 days. Cells were expanded over 2 weeks of time period. Phenotypes of CIK cells were analyzed by flow cytometric assays.
Cytotoxic assay
Cytotoxicity against MCF-7 and MCF-7/R cells was assessed by MTT (3-(4,5-dimethyl-2-thiazoyl)-2,5-diphenyl-2H-tetrazolium bromide) assay. Target cells were incubated with effector cells at various effector/target (E/T) ratios (10:1, 20:1, and 40:1) for 4 h. Target cells were used as a control to assess spontaneous mortality. Killing efficiency was calculated as follows: (experimental counts − effector spontaneous counts − target spontaneous counts) / (target maximal counts − target spontaneous counts) × 100. All the experiments were performed in triplicate.
MTT assay
MCF-7 and MCF-7/R cells treated with CIK cells at an E/T ratio of 20:1 for 12 h were washed three times with phosphate-buffered saline (PBS) to remove the suspended cells as previously described. 13 Then, breast cancer cells were inoculated into 96-well plates and incubated for five days counting from day 1. At the indicated time point, MTT solution was added into each well for 4 h. The formazan produced in each well by cells from MTT solution was solubilized by adding 200 µL dimethyl sulfoxide (DMSO). The absorbance at 490 nm was measured with a microplate reader.
To determine the effect of the soluble proteins secreted by CIK cells on the proliferation of MCF-7 and MCF-7/R cells, breast cancer cells were cocultured with CIK cells using a transwell chamber for 48 h. The membrane in the transwell had a 0.4-µm pore size that prevents both cell–cell contact and cell migration but allows the diffusion of soluble proteins. The cell proliferation was determined by MTT assay.
To evaluate the IC50 of 5-Fluorouracil (5-FU), docetaxel, and vinorelbine, the number of viable cells was determined at time 0 (control wells) and after 48 h treatment with EMEM alone (blank wells) or supplemented with chemotherapeutic agents. The absorbance was read at 490 nm using a microplate reader. The results were assessed using SPSS software (version 18; IBM Corp., Armonk, NY, USA) to calculate the IC50 (50% cell viability inhibition).
Invasion assay
Cell invasion assay was performed using a transwell assay. MCF-7 and MCF-7/R cells were treated with CIK cells at an E/T ratio of 20:1 for 12 h. Then, the cells were washed three times with PBS to remove the suspended cells. In total, 5 × 104 cells were placed in the upper chamber. The lower chamber was filled with 600 µL EMEM media supplemented with 20% FBS. The stained cells were counted under an inverted microscope (five fields per membrane). Each experiment was performed in triplicate.
Blocking studies
To investigate the mechanisms underlying the tumoricidal effect of CIK cells, CIK cells were first pre-incubated for 30 min with anti-human-NKG2D blocking antibody (BD Biosciences, San Jose, CA, USA) and isotype-matched antibody at 2.5 µg/mL, and subsequently cocultured with MCF-7 and MCF-7/R cells for 12 h, followed by CIK cell-mediated cytotoxicity.
To evaluate the contribution of MICA/B to kill target cells, MCF-7 and MCF-7/R cells were pre-incubated for 30 min with anti-MICA/B blocking antibodies (BD Biosciences) and isotype-matched antibody at 2.5 µg/mL before coculture assay.
Flow cytometry assay
The expression of MICA/B was evaluated on cells obtained from MCF-7 or MCF-7/R cells. In total, 5 × 105 cells were resuspended in 50 µL of PBS and labeled with anti-MICA/B and ULBP1–3 (BD Biosciences) and incubated for 20 min at room temperature. After incubation, the cells were washed twice and subsequently analyzed through flow cytometry.
MCF-7 and MCF-7/R cells were treated with CIK cells at an E/T ratio of 20:1 for 12 h. The cells were washed three times with PBS to remove the suspended cells. Apoptotic cells were analyzed using a staining kit (Sigma, St. Louis, MO, USA). The cells were incubated with Annexin-V-FITC and propidium iodide (PI) on ice for 30 min. Stained MCF-7 or MCF-7/R cells were analyzed by FACSCalibur flow cytometry using BD CellQuest software. Apoptotic cells are those stained with Annexin V-FITC+/PI− (early apoptotic cells) and Annexin V-FITC+/PI+ (late apoptotic cells).
Enzyme-linked immunosorbent assay
The coculture supernatants from CIK cells and tumor cells at an E/T ratio of 20:1 were measured using the Quantikine ELISA Kit (R&D Systems) according to the manufacturer’s instructions. The optical density of each sample was determined using a microplate reader at 450 nm, and the mean concentrations of IL-2, IFN-γ, IL-10, IL-4, and IL-6 were calculated.
Quantitative reverse transcription polymerase chain reaction
Total RNA was extracted from MCF-7 and MCF-7/R cells treated with or without CIK cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and the complementary DNA (cDNA) was synthesized by reverse transcription. Quantitative reverse transcription polymerase chain reaction (RT-PCR) was performed in duplicate on a LightCycler 2.0 Instrument (Roche Diagnostics, Mannheim, Germany) and analyzed using the comparative 2−ΔCT method as previously described. 14 Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control. The primers for TK1, TYMS, MDR1, and GAPDH are listed in Supplementary Table 1.
Western blot
MCF-7 and MCF-7/R cells treated with or without CIK cells were lysed in radioimmunoprecipitation assay (RIPA) buffer containing a protease inhibitor cocktail. Cell lysates were subjected to 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane. After blocking with TBST (Tris-buffered saline, 0.1% Tween 20) containing 5% non-fat dried milk, the membranes were incubated with the indicated antibodies, including anti-TK1, anti-TYMS, and anti-MDR1 (Abcam, Cambridge, MA, USA), overnight at 4°C, followed by washing with TBST and incubation with horseradish peroxidase (HRP) anti-mouse or anti-rabbit IgG for 1 h. The protein bands were detected with enhanced chemiluminescence.
Statistical analysis
The data are expressed as the mean ± standard error of the mean (SEM) or standard deviation (SD) based on triplet experiments. The data were analyzed using analysis of variance and Student’s t-test (two-tailed); p values <0.05 were considered statistically significant.
Results
Antitumor activity of CIK cells against breast cancer cells
CIK cells were generated by in vitro expansion of PBMCs from healthy donors using IFN-γ, anti-CD3 antibody, and IL-2. Flow cytometric analysis before and after 14 days of culture showed that the percentage of CD3+CD56+ cell subset increases from 3.776 ± 1.709 to 25.31 ± 7.42 (Supplementary Table 2). Cytotoxicity of CIK cells against MCF-7 or MCF-7/R cells was evaluated by cytotoxic assay. As shown in Figure 1(a), CIK cells were able to exert more significant cytotoxicity on MCF-7/R cells than MCF-7 cells.
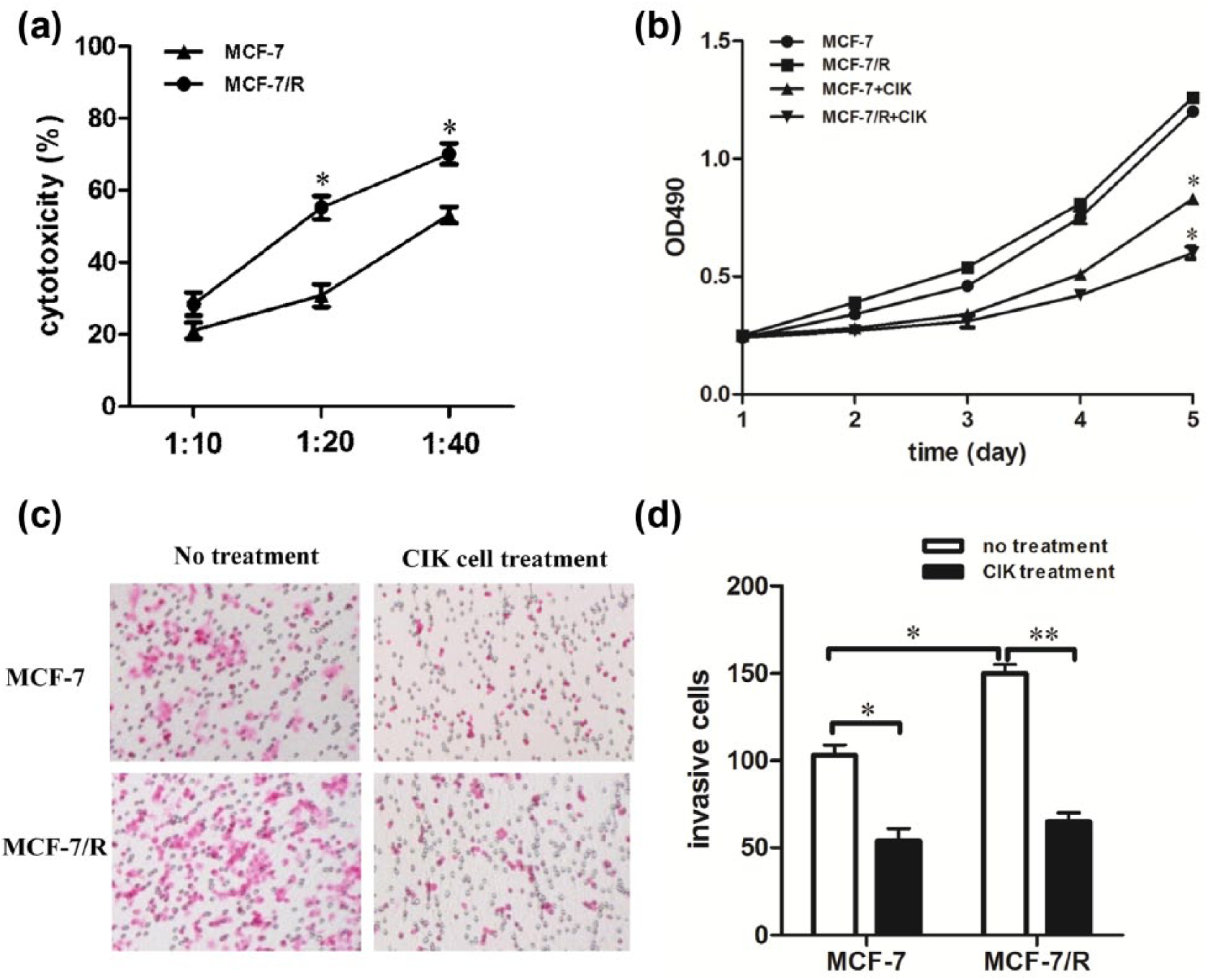
Radioresistant cells were more sensitized to CIK-mediated cytotoxicity. (a) Cytotoxicity percentages of CIK cells against MCF-7 and MCF-7/R cells at E/T ratios of 10:1, 20:1, and 40:1. (b) The cells were treated with CIK cells at an E/T ratio of 20:1 for 12 h. MCF-7/R cells grew slower than MCF-7 cells (p < 0.05). (c and d) MCF-7 and MCF-7/R cells were treated with CIK cells for 12 h at an E/T ratio of 20:1. Transwell invasion assay was used to determine tumor cell invasion. CIK cell treatment significantly inhibited tumor cell invasion (p < 0.05). Every experiment was repeated three times.
MCF-7 and MCF-7/R cells were cocultured with CIK cells at an E/T ratio of 20:1 for 12 hours, respectively. The suspended cells were removed and adherent cells were used for a growth curve assay. As shown in Figure 1(b), the growth curves of MCF-7 and MCF-7/R cells treated by CIK cells were dramatically dropped. And CIK-treated MCF-7/R cells grew slower compared with MCF-7 cells treated by CIK cells. Together, these results indicate that CIK cell treatment is more effective in inhibiting the proliferation of radioresistant cells.
We further explored the effect of CIK cell treatment on tumor cell invasion. The results showed that the invasion of CIK-treated MCF-7/R and MCF-7 cells was significantly reduced. No significant differences between two groups were observed. These results suggest that CIK cell treatment can markedly reduce tumor cell invasion.
MICA/B expression sensitizes radioresistant cells to CIK-mediated killing
It has been reported that tumor cell killing by CIK cells critically relies on cell contact–dependent lysis through a natural killer (NK)-like pathway.15,16 NKG2D molecules on CIK cells are a major membrane receptor for the recognition of target cells.17,18 We first determined the effect of CIK cell treatment plus anti-NKG2D blocking antibody on cell cytotoxicity against MCF-7 and MCF-7/R cells. We found that blocking antibodies to NKG2D significantly attenuated CIK-mediated cytotoxicity (Figure 2(a)). But there was no observable difference in the cytotoxicity against MCF-7 and MCF-7/R cells by CIK cells pretreated with anti-NKG2D antibodies. These results suggest that the differences of CIK-mediated cytotoxicity against MCF-7 and MCF-7/R cells are dependent on NKG2D-ligand recognition.

MICA/B expression sensitizes radioresistant tumor cells to CIK cell killing. (a) MCF-7 and MCF-7/R cells were cocultured with CIK cells at an E/T ratio of 20:1 in the presence of blocking antibody to NKG2D. Cytotoxicity percentage with anti-NKG2D antibodies was significantly reduced compared with isotype control antibody (p < 0.05), whereas no significant difference in cytotoxicity percentage of MCF-7 and MCF-7/R cells with anti-NKG2D antibodies was observed (p > 0.05). (b) MCF-7/R cells expressed higher levels of MICA/B compared to MCF-7 cells. (c–e) MCF-7 and MCF-7/R cells were pretreated with anti-MICA/B antibodies for 30 min and cocultured with CIK cells for 12 h. (c) Cytotoxicity percentage, (d) cell proliferation rates, and (e) tumor invasion were observed. Every experiment was repeated three times.
NKG2D ligands are stress-induced molecules, which are significantly upregulated by γ-irradiation.19,20 Next, we assessed the expression of NKG2D-ligand MICA/B on MCF-7 and MCF-7/R cells. As shown in Figure 2(b), the expression of MICA/B on MCF-7 cells was significantly lower compared to MCF-7/R cells. To prove a direct role of MICA/B in killing of tumor cells, MICA/B’s impact on MCF-7 and MCF-7/R cells was blocked by anti-MICA/B antibodies. Anti-MICA/B blocking antibodies significantly but partially suppressed CIK cell cytotoxicity (Figure 2(c)). No significant difference in the cytotoxicity of CIK cells against MCF-7 and MCF-7/R cells pretreated with MICA/B blocking antibodies was observed. These results indicate that MICA/B expression leads to the sensitization of MCF-7/R cells to CIK-mediated killing.
Subsequently, we observed that addition of MICA/B antibodies partially but significantly restored CIK-treated remarkable reduction in tumor cell growth and invasion (Figure 2(d) and (e)), indicating that MICA/B blockade partially inhibited tumor cell growth and invasion.
The effect of soluble proteins secreted by CIK cells on the growth of breast cancer cells
Breast cancer cells were cocultured with CIK cells by no cell contact at E/T ratios of 0, 10:1, 20:1, and 40:1 for 48 h. Figure 3(a) demonstrated that growth inhibition by CIK cells occurred at all E/T ratios tested but increased at higher ratios. In addition, breast cancer cells were cocultured with CIK cells by no cell contact at an E/T ratio of 20:1 for 48 h. The cells were cultured for another 1, 2, 3, 4, and 5 days. A growth curve assay showed that CIK-treated MCF-7 and MCF-7/R cells grew significantly slower compared with untreated cells (Figure 3(b)). These results suggested that the soluble proteins secreted by CIK cells might play an important role in inhibiting tumor growth.
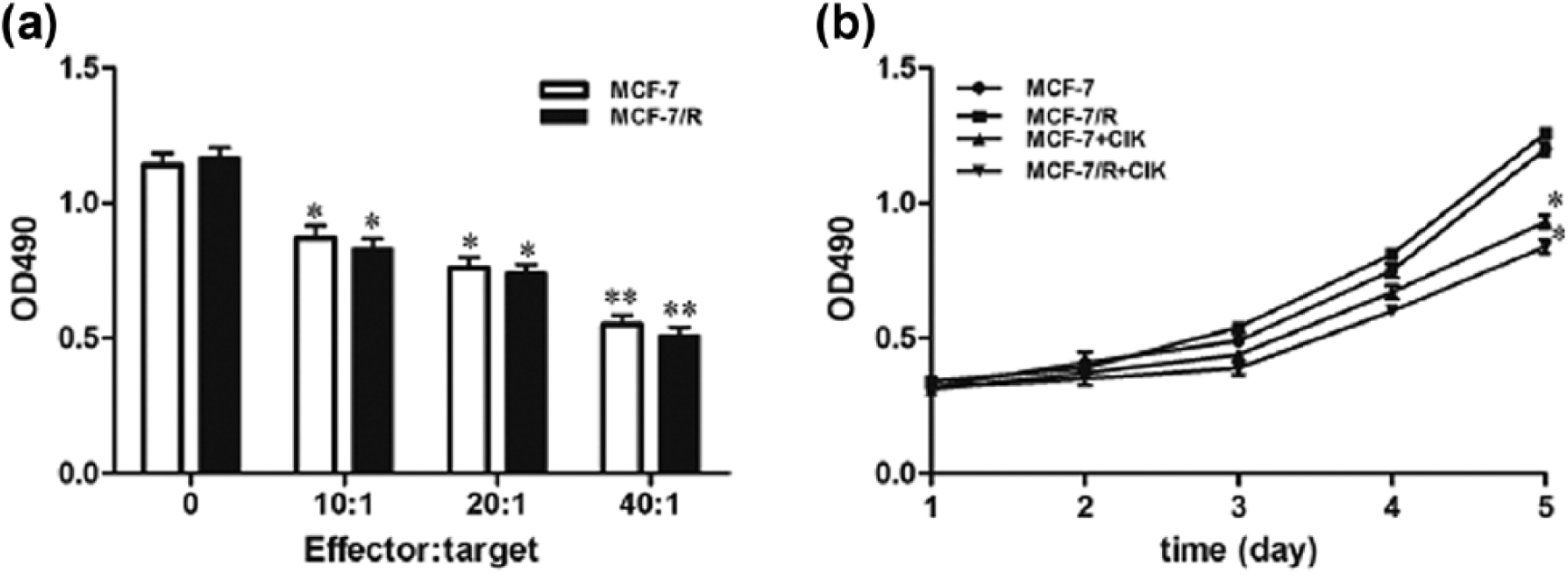
The effect of soluble proteins secreted by CIK cells on the proliferation of breast cancer cells. (a) Breast cancer cells were cocultured with CIK cells by no cell contact at different E/T ratios of 10:1, 20:1, and 40:1 for 48 h. CIK cell treatment significantly inhibited tumor cell growth. (b) Breast cancer cells were cocultured with CIK cells by no cell contact at an E/T ratio of 20:1 for 48 h. MCF-7 and MCF-7/R cells were cultured for another 1, 2, 3, 4, and 5 days. CIK cell treatment attenuated cell proliferation compared to no-treatment group (p < 0.05). Every experiment was repeated three times.
Cytokine production secreted by CIK cells in the coculture system
The proinflammatory cytokines secreted by CIK cells play a crucial role in inhibiting tumor cell growth.21,22 Thus, we tested the ability of CIK cells to secrete cytokines upon coculture with tumor cells at an E/T ratio of 20:1 for 48 h. As shown in Figure 4(a) and (b), IFN-γ and IL-6 were abundantly secreted by CIK cells, whereas IL-2, IL-4, and IL-10 levels had slight increase in the coculture system. Thus, once recognized, the proinflammatory cytokines IFN-γ and IL-6 might play an important role in CIK cell killing.

Cytokine productions secreted by CIK cells were evaluated in the coculture system. CIK cells were cocultured with MCF-7 and MCF-7/R cells at an E/T ratio of 20:1 for 48 h, respectively. (a and b) The cytokine levels (IFN-γ, IL-6, IL-2, IL-4, and IL-10) in the culture supernatant were determined by ELISA. Every experiment was repeated three times.
MCF-7/R cells treated with CIK cells exhibit increased apoptotic cells
Our aforementioned results revealed that MCF-7/R cells were more susceptible to CIK cell killing. The apoptosis of MCF-7 and MCF-7/R cells induced by CIK cells was further determined by flow cytometry. As shown in Figure 5 (a) and (b), the apoptotic rates of MCF-7/R cells were higher than MCF-7 cells. These results suggest that CIK cells have stronger antitumor activities against MCF-7/R cells via increasing cell apoptosis.

CIK-treated MCF-7/R cells exhibit increased apoptotic cells. (a) MCF-7 and MCF-7/R cells were cultured with CIK cells for 12 h and washed three times to remove suspended CIK cells. Tumor cells were stained with annexin V-FITC and PI labeling and analyzed by flow cytometry. (b) CIK-treated MCF-7 cells exhibited higher apoptotic rates compared to CIK-treated MCF-7/R cells (p < 0.05). Every experiment was repeated three times.
CIK cells sensitize breast cancer cells to chemotherapy
Chemotherapeutic agents such as 5-FU, docetaxel, and vinorelbine have been demonstrated to be effective in the treatment of breast cancer. To examine whether radioresistant cells were sensitive to chemotherapy, we assessed the drug resistance properties by MTT assay. As shown in Figure 6(a)–(c), 5-FU has similar IC50 for both MCF-7 and MCF-7/R cells. However, the IC50 values of docetaxel and vinorelbine in MCF-7 cells were significantly higher than that in MCF-7/R cells. These results suggest that MCF-7/R cells are more sensitive to chemotherapy as compared with MCF-7 cells.
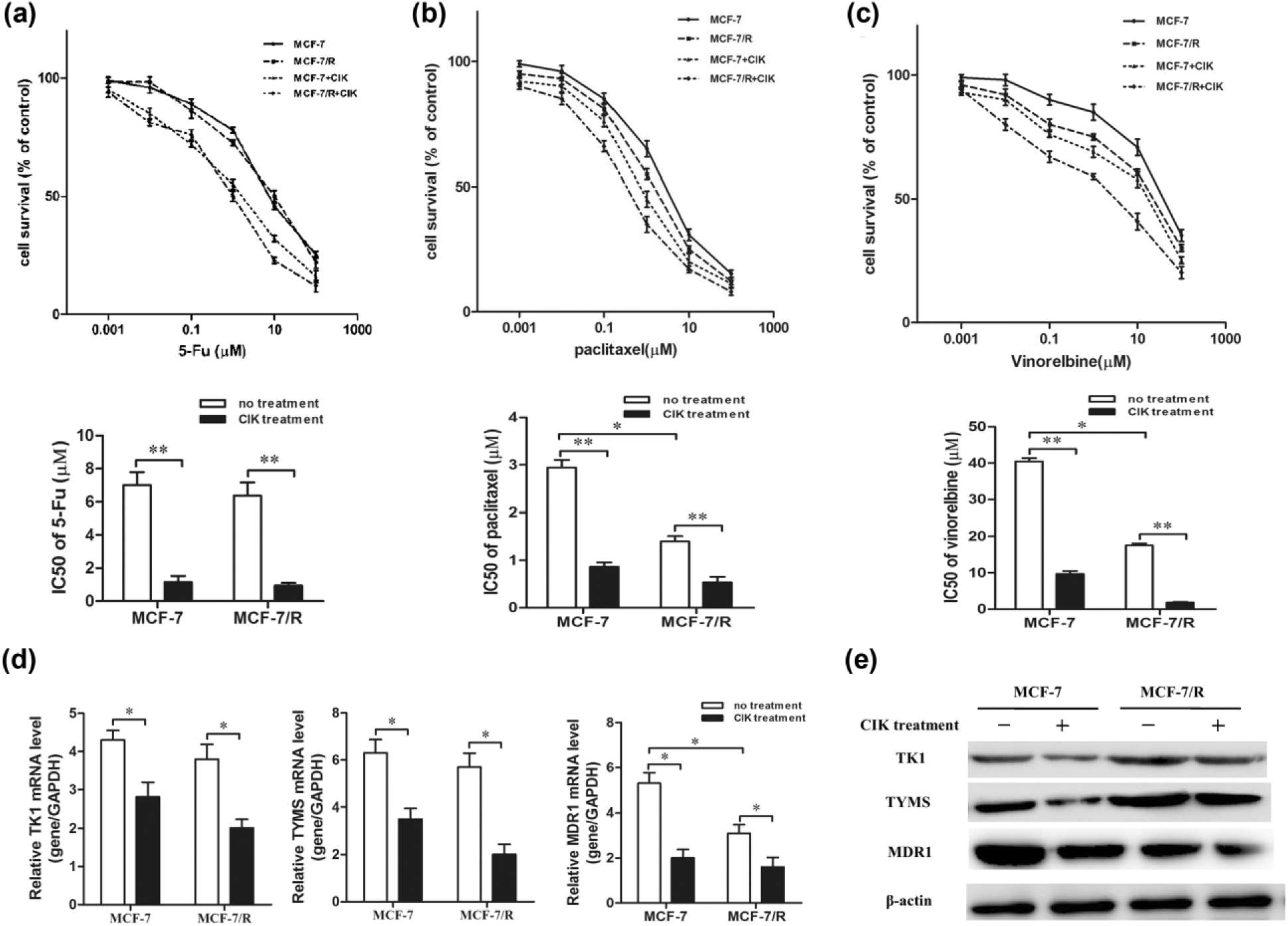
CIK cells sensitize breast cancer cells to chemotherapy. (a–c) The cells were treated with various concentrations of 5-FU, docetaxel, and vinorelbine for 48 h and subjected to MTT assay. CIK cell treatment reduced the IC50 of 5-FU, docetaxel, and vinorelbine in pairs of MCF-7 or MCF-7/R. (d–e) The mRNA and protein expression of TK1, TYMS, and MDR1 were analyzed by (d) qPCR and (e) western blotting. Every experiment was repeated three times.
Many clinical studies have reported that CIK cell treatment can enhance antitumor efficacy of chemotherapeutic agents.23,24 As shown in Figure 6(a)–(c), CIK cell treatment significantly decreased the IC50 of 5-FU, docetaxel, and vinorelbine, indicating that CIK cell treatment is able to sensitize breast cancer cells to chemotherapeutic agents.
It is reported that the expression of TK1, TYMS, and MDR1 is determinant of the sensitivity of 5-FU, docetaxel, and vinorelbine.25,26 To explore the mechanism of drug sensitization, we tested the effect of CIK cell treatment on the expression of TK1, TYMS, and MDR1. As shown in Figure 6(d) and (e), CIK treatment significantly reduced the messenger RNA (mRNA) and protein expression of TK1, TYMS, and MDR1 in MCF-7 and MCF-7/R cells. These results suggest that CIK cells could sensitize breast cancer cells to chemotherapeutic agents via the downregulation of TK1, TYMS, and MDR1.
Discussion
Patients with locally advanced cancer frequently receive prolonged treatment with fractionated radiotherapy. 27 Despite the initial clinical response, such effects are usually transient and relapse of most metastatic tumor frequently occurs due to the development of more resistant malignant cells to radiotherapy.28,29 Therefore, novel treatments aiming at targeting resistant cells to radiotherapy could find use in treating both primary and metastatic tumors.
CIK cells are a heterogeneous population of immune effector cells and show a wide MHC-unrestricted antitumor activity against different tumor cells. Other studies showed that combination of radiotherapy with CIK cell transfusion can improve clinical efficacy.30,31 However, such trials lack direct evidence of antitumor activity of CIK cells against radioresistant cells.
In this study, MCF-7/R cells with the enhanced aggressive phenotype were used as target cells for CIK cells. We found that CIK cells exhibited a stronger cytotoxicity against MCF-7/R cells and significantly inhibited MCF-7/R cell growth compared to MCF-7 cells. Taken together, these data demonstrate the more intense antitumor activity of CIK cells against radioresistant cells.
We further investigated the mechanism underlying the more efficient killing of radioresistant cells by CIK cells. Tumor cell recognition and cytotoxic activity by CIK cells are mainly mediated by the interaction of NKG2D with MICA/B and ULBPs. 32 In this study, we revealed that the direct antitumor activities of CIK cells against MCF-7 and MCF-7/R cells were dependent on NKG2D-ligand recognition. MICA/B as ligands for NKG2D is critical for the susceptibility of target cells to CIK cells in vitro and in vivo.33,34 Our data show that MCF-7/R cells express higher levels of MICA/B than MCF-7 cells. And CIK cells have similar antitumor effect on MCF-7/R and MCF-7 cells pretreated by the blocking antibodies to MICA/B. Therefore, these data, in part, support the idea that overexpression of MICA/B sensitized radioresistant cells to CIK cell killing.
In addition, our results showed that the soluble proteins secreted by CIK cells inhibited tumor cell proliferation. We further observed that CIK cells cocultured with MCF-7 and MCF-7/R cells produce significant amounts of IFN-γ and IL-6, and slight amounts of IL-2, IL-4, and IL-10, indicating that IFN-γ and IL-6 might play a critical role in killing tumors by CIK cells.
Increasing evidence supports the concept that the tumor bulk is consisted of heterogeneous populations which are resistant to radiotherapy and/or chemotherapy.35,36 Here, we find that MCF-7/R cells are more sensitive to chemotherapy compared to MCF-7 cells. It is known that CIK cell transfusion in combination with chemotherapy can improve clinical efficacy. 23 We observed that breast cancer cells become more sensitive to chemotherapy after cocultured with CIK cells. Next, the mechanism of sensitivity of CIK-treated breast cancer cells to chemotherapy was investigated. We found that CIK cell treatment had inhibitory effects on TK1, TYMS, and MDR1 expression, which is involved in drug resistance. Together, these data provide direct evidence that CIK immunotherapy sensitizes tumor cells to chemotherapy.
In summary, our findings here demonstrate intense antitumor activity of CIK cells against radioresistant cells. However, CIK cells are able to sensitize radioresistant cells to chemotherapy, supporting the idea that immunotherapy in synergism with other therapeutic strategies (e.g. chemotherapy and/or radiotherapy) improves clinical efficacy.
Footnotes
Acknowledgements
Q.G., D.Z., and X.B. contributed equally to this study.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81501422) and Qingdao Outstanding Health Professional Development Fund.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.