
Abstract
Multidrug resistance in tumor cells is still a big challenge in cancer treatment. Therefore, identification ofsafe and effective multidrug resistance–reversing compounds with minimal side effects is an important approach in cancer treatment. Here, we investigated the role and potential mechanisms of peroxisome proliferator–activated receptor γ in doxorubicin-resistant human myelogenous leukemia (K562/DOX) cells. The effect of doxorubicin on cell viability following treatment with balaglitazone, a peroxisome proliferator–activated receptor γ agonist, was evaluated using trypan blue and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assays. Rhodamine123 assay was used to determine the activity of two common drug efflux membrane transporters P-glycoprotein and multidrug resistance protein-1. P-glycoprotein, multidrug resistance protein-1, and phosphatase and tensin homolog deleted on chromosome 10 messenger RNA/protein expression levels were measured by quantitative reverse transcription polymerase chain reaction and western blot analyses. Annexin V/fluorescein isothiocyanate assay was also employed to investigate apoptosis. We showed that balaglitazone considerably enhanced the cytotoxicity of doxorubicin. Balaglitazone also significantly downregulated P-glycoprotein expression and activity in K562/DOX cells and reduced multidrug resistance through elevation of intracellular doxorubicin in cells. Furthermore, upon balaglitazone treatment, phosphatase and tensin homolog deleted on chromosome 10 expression could be restored in K562/DOX cells in a peroxisome proliferator–activated receptor γ–dependent manner. We concluded that peroxisome proliferator–activated receptor γ agonist, balaglitazone, could reverse multidrug resistance by inducing phosphatase and tensin homolog deleted on chromosome 10 and peroxisome proliferator–activated receptor/ phosphatase and tensin homolog deleted on chromosome 10 signaling pathway. These findings suggest that targeting peroxisome proliferator–activated receptor γ might serve as an effective approach for circumventing multidrug resistance in chemotherapy of cancerous patients.
Keywords
Introduction
Chemotherapy is still the main strategy for cancer treatment. However, a majority of patients suffer from tumor relapse due to chemoresistance. Development of multidrug resistance (MDR) is mainly responsible for the lack of sensitivity in tumor cells by reducing intracellular drug accumulation through enhancing drug efflux. MDR is generally associated with the overexpression and/or hyper-activation of membrane adenosine triphosphate (ATP)-binding cassette (ABC) transporters that regulate the intracellular concentrations of many cytotoxic drugs and molecularly target cancer therapeutics.1–3
P-glycoprotein (P-gp), encoded by the MDR-1 gene, and multidrug resistance protein-1 (MRP-1), encoded by the ABCC1 gene, are dominant members of ABC transporters in myeloid leukemia and can efflux different types of chemotherapeutic agents such as anthracyclines, taxanes, and vinca alkaloids.4,5 Therefore, inhibition of P-gp and MRP-1 has been suggested as a potential strategy to overcome resistance in myeloid leukemia. Different types of P-gp or MRP-1 inhibitors such as verapamil and cyclosporin A have been combined with anticancer agents to circumvent MDR. 6 However, these compounds failed to be clinically approved due to their cardiotoxicity, adverse side effects, and pharmacokinetic interactions. Therefore, identification of effective MDR-reversing compounds with minimal adverse effects is an attractive research frontier in many laboratories around the world.
Recent cancer therapy approaches are focusing on targeting the mediators that inhibit specific pathways that contribute to the malignant and/or resistance phenotype of cancer cells. Phosphoinositide 3-kinase (PI3K)/Akt signaling pathway regulates many processes important for cancer development, for example, cell proliferation, cell survival, angiogenesis, and metastasis. 7 Studies have shown that resistance to leukemia chemotherapy is correlated with the activation of PI3K/Akt signaling pathway, as manifested by the fact that inhibition of this signaling pathway can reverse MDR and sensitize tumor cells to chemotherapeutics.7–9
Peroxisome proliferator–activated receptors (PPARs) are nuclear steroid receptors that regulate diverse biological processes such as lipid and carbohydrate metabolism, development, differentiation, apoptosis, neoplastic transformation, inflammation, and regeneration.10–12 PPARs form heterodimers with retinoid X receptors (RXRs) and bind to PPAR response elements (PPREs) located in the promoter of target genes to regulate transcription.13,14 PPARγ agonists have also been demonstrated to have beneficial effects in different types of tumor cells including gastric, 15 colon, 16 leukemia, 17 hepatocellular carcinoma, 18 pancreas, 19 breast, 20 and prostate 21 cancers. PPARγ agonists have been reported to activate phosphatase and tensin homolog deleted on chromosome 10 (PTEN) protein expression. 22 PTEN is a tumor suppressor protein and inhibits the activation of PI3K/Akt cascade via dephosphorylation of position 3 phosphate of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) to produce phosphatidylinositol 4,5-bisphosphate (PIP2); thereby, downregulation of PTEN in different kinds of solid tumors and hematopoietic malignancies may be correlated with poor tumor prognosis and drug resistance.23,24
Several recent studies have reported that targeting PPARγ might be an effective adjunct therapy in leukemia. In a very recent study, Prost et al. 25 demonstrated that PPARγ agonists synergistically reduced the number of colony-forming cells in patients with chronic myeloid leukemia (CML). Lin et al. 26 showed that the expression levels of ABC transporters and PTEN in tumor cells are important determinants of combination therapy efficacy and proposed that PTEN status of the tumor should be taken into account during the analysis of the clinical trials. All these finding led us to hypothesize that PPAR agonists may reverse MDR in cancer cells via PTEN regulation.
In this study, we aimed to examine the role of PPARγ, alone and combination with doxorubicin (DOX) treatment, on viability, apoptosis, and MDR reversing of the DOX-resistant human myelogenous leukemia (K562/DOX) cells. Balaglitazone was used as a PPARγ agonist to test the hypothesis. DOX, a substrate for P-gp and MRP-1, is one of the chemotherapeutic drugs used in the treatment of myeloid leukemia. To uncover the potential molecular mechanism, P-gp, MRP-1, and PTEN messenger RNA (mRNA)/protein expression levels as well as P-gp and MRP-1 activities were investigated in the presence or absence of different signaling pathway inhibitors.
Materials and methods
Human CML (K562) and DOX-resistant human CML (K562/DOX) cells were obtained from Pasteur Institute Cell Culture Collection (Tehran, Iran); DOX and LY294002 (a PI3K/Akt pathway inhibitor), PD098059 (a mitogen-activated protein kinase (MAPK) pathway inhibitor), GW9662 (a PPARγ antagonist), verapamil (a P-gp inhibitor), and MK571 (a MRP-1 inhibitor) were purchased from Cayman (USA). SF1670 (a PTEN inhibitor) was obtained from Echelon Biosciences Inc, (USA). Rhodamine123 (Rh123), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), propidium iodide (PI), and RNase were purchased from Sigma (USA). Fetal bovine serum (FBS) and RPMI-1640 medium were purchased from Gibco (USA). Antibodies for P-gp, MRP-1, PTEN, and β-actin were obtained from Abcam (Cambridge, UK).
Cell culture
K562 and K562/DOX cells were cultured in RPMI-1640 medium supplemented with 10% FBS at 37°C in a humidified atmosphere with 5% CO2. K562/DOX cells were cultured in the presence of 0.5 μM of DOX and were grown in drug-free medium for 2 weeks before being used in experiments.
Cell viability assay
MTT assay was used for cell viability analyses, as described previously. 27 This method is based upon the reduction of yellow MTT to insoluble formazan in the mitochondria of viable cells. Briefly, K562 and K562/DOX cells were seeded in a 96-well plate in RPMI-1640 medium supplemented with 10% FBS at the density of 2 × 104 cells/well. After 24 h incubation, various concentrations of DOX with or without balaglitazone were diluted in RPMI-1640 medium (without FBS) and added into each well. Experiments for each group were performed in triplicates and with a blank control. After 48 h of treatment, the medium was removed and 200 μL of RPMI-1640 medium supplemented with 10% FBS and 10% MTT (5 mg/mL) was added. After incubation for another 4 h, the reduced intracellular formazan product was dissolved by replacing 100 μL of RPMI-1640 medium with the same volume of dimethyl sulfoxide (DMSO). Absorbance values were measured at 570 nm with a micro plate reader (State Fax 2100; Awareness Technology Inc, USA). The half maximal inhibitory concentration (IC50) of each experiment was calculated. The resistance fold (RF) was calculated by dividing the IC50 value of treatment in resistant cells by the IC50 value of treatment in corresponding parental cells.
Rho123 uptake and accumulation assays
In the uptake assay, cells were cultured in a six-well plate at the density of 1 × 106 cells/well. After 24 h of incubation, cells were treated with medium containing 5 μM Rho123 in the presence or absence of 25 μM of balaglitazone and were incubated at 37°C for 30, 60, 90, and 120 min. Verapamil and MK571 were used as positive controls for P-gp and MRP-1 inhibitors, respectively. For Rho123 accumulation assay, K562 and K562/DOX cells were cultured in a six-well plate. Cells were incubated in the presence or absence of 25 μM of balaglitazone with medium containing 5 μM of Rho123 at 37°C for 2 h. The intracellular mean fluorescence intensity (MFI) associated with Rho123 was measured using a FP-6200 fluorometer (Jasco Co., Tokyo, Japan). Excitation was performed by an argon ion laser operating at 488 nm, and the emitted fluorescence was collected through a 530 nm pass filter.
RNA isolation, complementary DNA synthesis, and real-time reverse transcription polymerase chain reaction
Total cellular RNA was isolated from cells using the AccuZolTM reagent following the manufacturer’s instructions (Bioneer; Daedeok-gu, Daejeon, Korea). Total RNA (2 μg) was subjected to the reverse transcription (RT) reaction using Moloney Murine Leukemia Virus (MMLV) reverse transcriptase (Promega, USA) and oligo-dT primer. The complementary DNA (cDNA) products were further subjected to polymerase chain reaction (PCR) amplification with SYBR Premix Ex Taq (TaKaRa Bio, Otsu, Japan) and specific primers using the Rotor-Gene™ 6000 system (Corbett Life Science, Mortlake, Australia). All experiments were carried out in triplicates, and for each sample assayed, the threshold cycle (Ct) value for target gene or β-actin gene was determined. The relative mRNA expression levels of each gene were normalized to the β-actin expression level, which allowed target cDNA calculation as 2−(Ct target gene − Ct β-actin).
Western blotting analysis
Western blots were performed according to Mohammadzadeh et al. with slight modifications. 28 Cell lysates were collected using a buffer (20 mM Tris-HCl at pH 7.5) containing detergents (0.1% sodium dodecyl sulfate (SDS), 1% sodium deoxycholate, and 1% Triton X-100). The protein concentration in the cell lysates was measured by Bradford reagent with bovine serum albumin (BSA) as standard (Bio-Rad, USA). Each 50 μg of lysate was resolved by corresponding SDS–polyacrylamide gel electrophoresis (PAGE) with different percentages and then transferred onto polyvinylidene difluoride (PVDF) membranes. After blocking with milk, the membranes were incubated with primary antibodies for overnight at 4°C. The membranes were then washed with Tris-buffered saline with 0.1% Tween 20 (TBST) buffer three times and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody at room temperature for 2 h. Subsequently, the membranes were washed three more times with TBST buffer and were developed by the enhanced chemiluminescence (ECL) reagents (Amersham Pharmacia Biotech, USA).
Flow cytometric apoptosis assay
The K562/DOX cells at a density of 5 × 105/mL in exponential growth were exposed to 0.5 μM of DOX in the presence or absence of balaglitazone and were washed twice with the phosphate-buffered saline (PBS) after 24 h. The cell pellets were resuspended in 100 µL of PI and fluorescein isothiocyanate (FITC)-labeled Annexin V solution for 10 min in room temperature and then analyzed using a fluorescence-activated cell sorting (FACS) flow cytometer (BD LSR; Becton-Dickinson, USA) and Cell Quest software (Becton-Dickinson).
Statistical analysis
Data were presented as mean ± standard deviation (SD). Analysis of variance (ANOVA) followed by Bonferroni’s and Sidak’s tests were used to determine the significant differences between groups. p values less than 0.05 were considered to be significant. All statistical analyses were performed using GraphPad Prism 6.01 software.
Results
Effects of balaglitazone on the viability of K562 and K562/DOX cells
The effect of balaglitazone on cell viability was determined with MTT assay. As shown in Figure 1, balaglitazone (concentration range from 5 to 100 μM) displayed equal cytotoxicity towards the MDR cells as well as the corresponding parental cells. The IC50 values for DOX were 0.121 μM in K562 and 1.95 μM in K562/DOX cells, the latter showing 16.11-fold resistance to DOX, compared to the parental K562 cells.

The sensitivity of K562 and K562/DOX cells to DOX and balaglitazone. Cells were treated with various concentrations of (a) DOX and (b) balaglitazone for 48 h; cell viability was determined by the MTT assay. Each point represents the mean ± SD (n = 3 independent experiments; *p < 0.05 vs K562/DOX cells).
Balaglitazone reversed MDR in K562/DOX
As shown in Figure 1, balaglitazone had no significant inhibitory effects on the growth of K562 and K562/DOX cells at the concentrations ranging from 5 to 40 μM, while the anti-proliferative effects was observed at higher concentrations (50–100 μM). To minimize the effect of balaglitazone itself on the resistant cell growth, we selected a lower concentration than IC50 values, 25 μM, in the MDR reversal experiments. The effect of balaglitazone on the sensitivity of K562 and K562/DOX cells to DOX has been shown in Figure 2. Left shifts in the cytotoxicity profiles of DOX upon the addition of balaglitazone were indicative of MDR reversal in K562/DOX cells. The treatment of balaglitazone at non-toxic concentrations induced a significant decrease in DOX IC50 against K562/DOX cells (Table 1). Therefore, a more significant difference in the RF value (4.36) was seen in K562/DOX cells for balaglitazone (p value < 0.05).
Effect of exposure to balaglitazone on doxorubicin cytotoxicity in K562 and K562/DOX cells.

IC50: half maximal inhibitory concentration; RF: resistance fold.
Resistant cells are maintained in doxorubicin-free medium for 1 week before seeding for cell viability assay and measured as described in Materials and methods section. Each value represents the mean ± SD of three independent experiments. The RF was determined as the ratio of the IC50 value of the resistant cells to that of the corresponding sensitive parental cells.
Significance at p < 0.05.

Effects of balaglitazone co-treatment on enhancing DOX cytotoxicity in K562 and K562/DOX cells. (a) K562 cells and (b) K562/DOX cells were treated with DOX and DOX plus 25 µM of balaglitazone for 48 h, and the cell viability was determined by the MTT assay. Each point represents the mean ± SD (n = 3 independent experiments; *p < 0.05 vs control).
Effects of balaglitazone on P-gp and MRP-1 activities
To investigate whether the PPARγ agonist effects were mediated by P-gp and/or MRP-1, the intracellular Rh123-associated MFI was examined in parental K562 and K562/DOX cells, to determine the activity levels of P-gp and MRP-1. Verapamil and MK571 were used as positive controls. As shown in Figure 3(a), in the uptake study, after K562/DOX cells were incubated with Rh123 (substrate of P-gp and MRP-1) in the presence of balaglitazone, the MFI increased for balaglitazone in a time-dependent manner over a 2 h period (p value < 0.05) and was comparable to that in verapamil-treated K562/DOX cells; however, in other groups, the substrate did not accumulate over the same duration. Figure 3(b) clearly illustrates that balaglitazone enhanced Rh123 accumulation in K562/DOX cells after 2 h; in contrast, no such increase in MFI was observed in balaglitazone-treated K562 cells. All these findings suggested that balaglitazone may inhibit the activity of P-gp and/or MRP-1 in K562/DOX cells.

Effect of balaglitazone on the uptake and intracellular accumulation of Rh123. (a) K562/DOX cells were incubated with medium containing 5 µM of Rh123 in the presence or absence of 25 µM of balaglitazone at 37°C for 30, 60, 90, and 120 min, and MFI was measured as described in the Materials and methods section. (b) Cells were incubated with medium containing 5 µM of Rh123 in the presence or absence of 25 µM of balaglitazone, 10 µM of verapamil, and 10 µM of MK571 at 37°C for 2 h, and then, Rh123-associated MFI was measured. Each point represents the mean ± SD from four experiments (*p < 0.05 vs control).
Balaglitazone downregulated P-gp expression in K562/DOX cells
We first evaluated the protein levels of P-gp and MRP-1 in K562 and K562/DOX cells. The findings showed that parental K562 cells significantly expressed a low basal level of the P-gp transporter, but MRP-1 expression was not different between two cell groups (Figure 4). We found out that balaglitazone downregulated MDR-1 and P-gp in the mRNA and protein levels in K562/DOX cells, which can explain the enhanced intracellular accumulation of DOX and cytotoxicity in the balaglitazone-treated K562/DOX cells (Figure 5(a) and (b)).
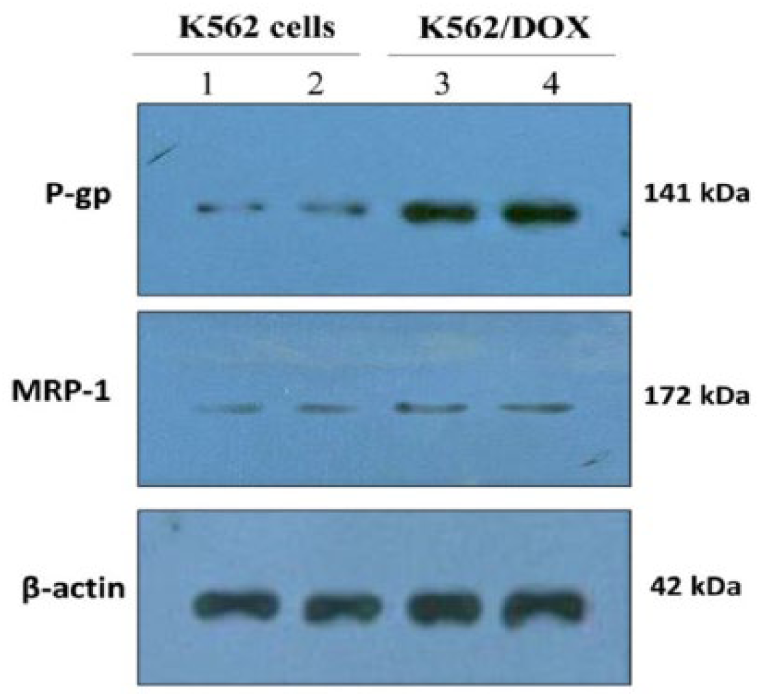
The basal levels of P-gp and MRP-1 in K562 and K562/DOX cells. Lane 1 and 2: K562 cells; lane 3 and 4: K562/DOX cells. β-actin protein was used as a loading control.

Effects of balaglitazone on P-gp and MRP-1 expression. K562/DOX cells were cultured in the presence or absence of balaglitazone (25 µM) for 48 h. (a) Protein expression P-gp, MRP-1, and β-actin was examined by western blot analyses. (b) MDR-1 mRNA levels were determined by qRT-PCR, and relative mRNA expression was quantified by the 2 (−ΔΔCt) method. The data represent mean ± SD (n = 3; *p < 0.05 vs K562 cells; #p < 0.05 vs untreated K562/DOX cells).
Effect of balaglitazone on MDR in presence of LY294002, PD098059, and SF1670
To further investigate the underlying molecular mechanisms, we examined the MDR-reversing effect of balaglitazone in three concentrations and also in presence of specific inhibitors of PI3K (LY294002), MAPK (PD098059), and PTEN (SF1670). As shown in Table 2, compared with the control, DOX cytotoxicity was enhanced in the balaglitazone-treated group. The IC50 values (μM) were decreased from 1.95 ± 0.17 to 0.96 ± 0.08, 0.53 ± 0.09, and 0.40 ± 0.05. However, balaglitazone effect was significantly attenuated by SF1670 treatment.
Effect of the single balaglitazone and co-exposure with LY294002, PD098059, and SF1670 on the cytotoxicity of DOX and reversing MDR in K562 and K52/DOX cells.
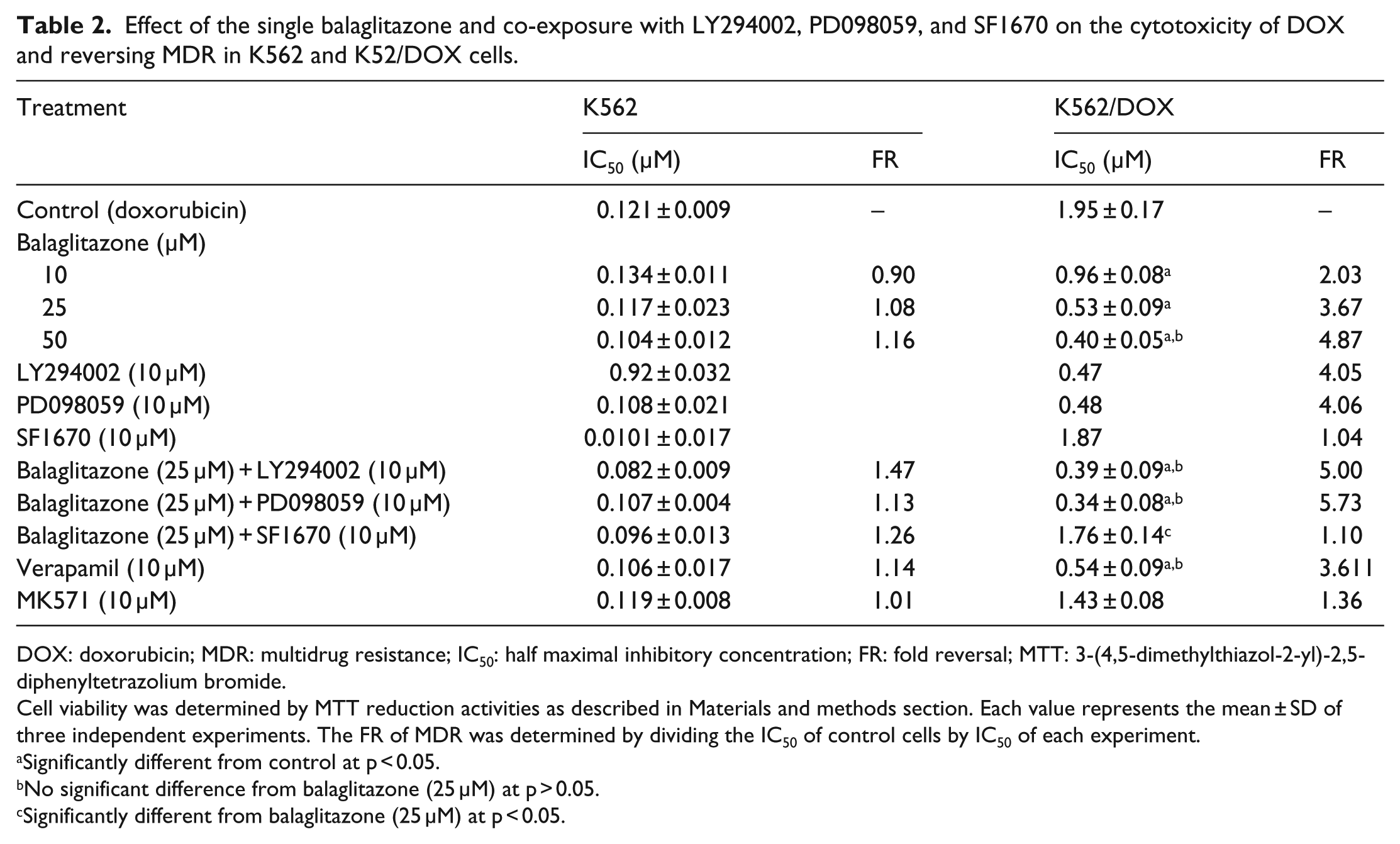
DOX: doxorubicin; MDR: multidrug resistance; IC50: half maximal inhibitory concentration; FR: fold reversal; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
Cell viability was determined by MTT reduction activities as described in Materials and methods section. Each value represents the mean ± SD of three independent experiments. The FR of MDR was determined by dividing the IC50 of control cells by IC50 of each experiment.
Significantly different from control at p < 0.05.
No significant difference from balaglitazone (25 μM) at p > 0.05.
Significantly different from balaglitazone (25 μM) at p < 0.05.
PTEN inhibition abolished balaglitazone effects on P-gp functionality
As shown in Figure 6, after incubating the cells with 10, 25, and 50 μM of balaglitazone for about 2 h, the concentration-dependent effect of the balaglitazone on the intracellular Rh123-associated MFI was observed in K562/DOX cells; balaglitazone effect was not significant in K562 cells. However, the effect of 25 μM of balaglitazone in K562/DOX cells was attenuated by 10 μM of SF1670 co-treatment. Furthermore, inhibition of MRP-1 activity by 10 μM of MK571, a specific MRP-1 activity inhibitor, showed that MRP-1 was not involved in DOX resistance in K562/DOX cells.
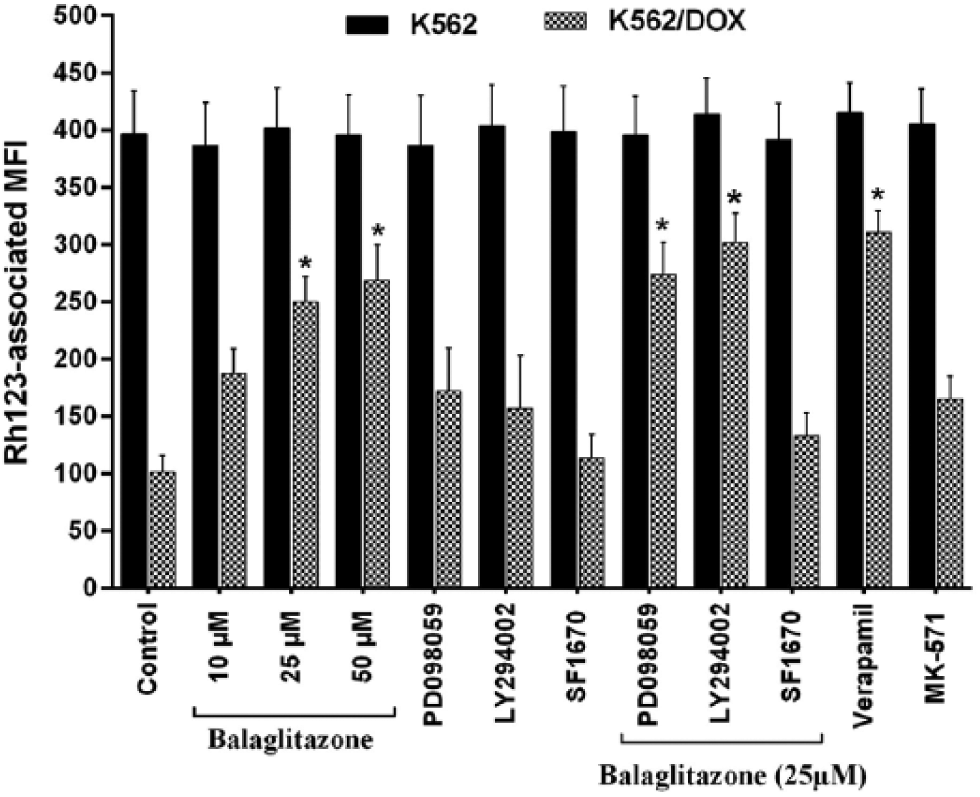
Effect of LY294002, PD098059, and SF1670 on the balaglitazone-induced intracellular Rh123 accumulation in K562 and K562/DOX cells. After incubation for 2 h, balaglitazone elevated Rh123-associated MFI in a concentration-dependent manner in K562/DOX cells but had no effect on that in K562 cells. Co-treatment with 10 µM of LY294002 or PD098059 increased Rh123-associated MFI and with 10 µM of SF1670 almost abolished the action of the balaglitazone. Each bar represents the mean ± SD (n = 4) and was measured as described in Materials and methods section (*p < 0.05 vs control).
Balaglitazone downregulated P-gp via upregulation of PTEN in K562/DOX cells in a PPARγ-dependent manner
PTEN is a tumor suppressor protein, which induces cell death and MDR reversal effect by inhibition of PI3K/Akt signaling pathway. This eventually leads to downregulation of P-gp and increase in intracellular drug accumulation. In order to evaluate the relationship between balaglitazone and PTEN, PTEN expression was evaluated in cells. Surprisingly, we detected that PTEN expression was significantly lower in K562/DOX cells (Figure 7(a) and (b)). The findings clearly demonstrated that balaglitazone upregulated PTEN expression after 48 h of incubation in a dose-dependent manner. In addition, further investigations revealed that co-incubation of K562/DOX cells with balaglitazone in presence of 10 μM of SF1670 (PPARγ antagonist) eliminated balaglitazone effects on P-gp expression, while no similar effect with 10 μM of LY294002 or 10 μM of PD098059 was noted (Figure 8(a) and (b)). Collectively, these results demonstrated that balaglitazone-induced downregulation of the P-gp was associated with PTEN expression in a PPARγ-dependent manner in K562/DOX cells.

Effect of balaglitazone on the PTEN protein expression level (a) and PTEN mRNA expression (b) in K562/DOX cells. Balaglitazone upregulated PTEN in K562/DOX cells in a dose-dependent manner; 25 µM of GW9662 abolished the action of the balaglitazone. The data represent mean ± SD (n = 3; #p < 0.05 vs parental K562 cells; *p < 0.05 vs untreated K562/DOX cells).

Effect of 25 μM of baglitazone, 10 μM of PD98059, 10 μM of LY29402, 10 μM of SF1670, and GW9662 on the P-gp expression (a) and MDR-1 mRNA (b) in K562/DOX cells. SF1670 and GW9662 suppress the activity of balaglitazone. Data represent mean ± SD (n = 3; *p < 0.05 vs untreated K562/DOX cells).
Balaglitazone promoted DOX-induced apoptosis in K562/DOX cells
The findings showed that the percentage of Annexin V

Flow cytometric analysis of K562/DOX cells. Cells were stained with Annexin V–FITC and PI after treatment with balaglitazone (25 µM), DOX (0.5 µM), and balaglitazone (25 µM) plus DOX (0.5 µM).
Discussion
MDR in tumor cells is a major burden in cancer treatment.2,3 Therefore, identification of safe and effective MDR-reversing compounds and mechanisms is an important goal in clinical research.
PPARγ, as the most studied member of PPARs family in cancer therapy, is a therapeutic target for potential drug discovery. 10 Research has shown that PPARγ ligands generally decrease the survival rate of cancer cells through induction of apoptosis and regulation of gene or protein expression levels associated with cell cycle arrest.29,30 Moreover, recent studies have demonstrated that balaglitazone, a PPARγ agonist, may prove useful in preventive medicine, and this effect may be PPARγ-dependent.31,32
The results of our study showed that balaglitazone causes a significant decrease in the viability of K562/DOX cells. In addition, the effects of balaglitazone were shown to reduce P-gp expression and to abolish MDR in cancer cells. Our results indicated that exposing resistant cells to different concentrations of balaglitazone can reverse MDR by increasing the intracellular accumulation of DOX, inhibiting the P-gp activity, and increasing PTEN expression in a PPARγ-dependent manner in K562/DOX cells.
Thus far, only a few studies have focused on the mechanisms by which PPARγ suppresses cancer cell proliferation.33 –35 In one such study, Cheon et al. 36 assessed the effects of ciglitazone and troglitazone on the proliferation of the human gastric cancer cells. They showed that PPARγ activation suppressed cell growth and that troglitazone was more potent than ciglitazone. The results of our study are in line with the above-mentioned studies. The intracellular mechanisms related to the decreased viability of the resistant cells remained unclear.
Although chemotherapy has created a long life expectancy for cancer patients, the treatment is associated with certain side effects. For instance, cardiotoxicity is one of the most dreadful side effects encountered in the case of various agents, in particular DOX.37,38 The incidence of cardiac toxicity largely depends on drug dose. Combining chemotherapeutics with other agents is a novel treatment strategy for decreasing the lethal side effects. 39 In this study, we showed that a combination of DOX and balaglitazone at non-toxic concentrations caused a significant decrease in the IC50 values of DOX in a concentration-dependent manner. Therefore, simultaneous treatment with DOX and balaglitazone reduced the DOX therapeutic dose and, more importantly, may relieve DOX-induced side effects without any changes in its efficacy.
In addition to increase in the potency of DOX against cancer cells, PPAR ligands are effective in reversing MDR. One of the most common causes of MDR is overexpression of P-gp. 5 P-gp is the direct target of studies in which PPAR ligands were used for overcoming MDR. 40 In one of these studies, Zhang et al. treated a MDR ovarian cancer cell subline with different concentrations of rosiglitazone, a PPARγ ligands. They showed that rosiglitazone downregulated P-gp expression in a concentration-dependent manner, possibly by downregulation of Wnt/β-catenin pathway through suppression of the FZD1. 41 In another study, rosiglitazone was used to potentially reverse mitomycin C resistance in human drug-resistant gastric cancer cell line. 42 The authors reported that rosiglitazone administration enhanced the apoptosis rate in a dose-dependent manner. In addition, rosiglitazone reversed mitomycin C resistance in cancer cells by reducing the expression levels of P-gp.
We have demonstrated similar results in our study. Treatment with balaglitazone significantly decreased the activity as well as the mRNA/protein expression levels of P-gp in K562/DOX cells. In other words, balaglitazone is capable of inhibiting the efflux of Rh123 from cells. On the contrary, in the presence of balaglitazone, DOX accumulation was increased in resistant cells, which is another explanation for enhancement of DOX cytotoxicity in balaglitazone-treated cells.
To investigate the role of balaglitazone in the chemoresistance of K562/DOX cells, we examined the effect of balaglitazone incubation on DOX-induced apoptosis. Flow cytometric results showed that DOX in combination with balaglitazone dramatically enhanced apoptosis, while we did not detect significant apoptosis in K562/DOX cells upon single balaglitazone or DOX treatment.
PTEN protein is a member of phosphatase family with dual lipid and protein phosphatase functions. Since PTEN negatively regulates PI3K/Akt signaling pathway, it has been postulated that it can inhibit proliferation, induce apoptosis, and modulate cell cycle progression.23,24 On the contrary, overexpression of PTEN has been associated with increased senility of tumor cells in response to chemotherapeutic agents. 43 In this regard, P-gp is believed to have a role in elevation of chemotherapy efficacy. In a study by Lin et al., 26 it was reported that the efficacy of combination therapy is absolutely dependent on the ABC transporters, in particular P-gp and breast cancer resistant protein (BCRP), as well as on the presence of PTEN and its status in tumor cells. They concluded that the PTEN evaluation in tumor should be taken into account during clinical trials. In another study, Lee et al. 22 showed that rosiglitazone potentiated the anti-proliferative activity of gefitinib by increasing PTEN expression levels in the non–small cell lung cancer. In agreement with the above study, our results surprisingly showed that PTEN mRNA levels were significantly lower in K562/DOX cells when compared with their parental cells. In addition, we found that balaglitazone induced PTEN expression in a concentration-dependent manner. Furthermore, combination treatment with balaglitazone and GW9662, as PPARγ antagonists, abolished the favorable effects of balaglitazone on PTEN expression. Therefore, balaglitazone decreased P-gp and increased PTEN expression in K562/DOX cells through PPARγ.
Taken together, our study suggested that targeting PPARγ with balaglitazone might be an effective approach for combination therapy in leukemia treatment and circumventing MDR against conventional chemotherapeutic drugs. Moreover, we found that overcoming MDR after treatment with balaglitazone resulted in PTEN increase and that PPAR/PTEN signaling pathway participates in the reversal process of P-gp-mediated resistance upon treatment with balaglitazone.
Footnotes
Acknowledgements
B.Y., N.Z., and N.S. contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.