
Abstract
Acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitors, such as gefitinib and erlotinib, is a critical issue in the treatment of patients with epidermal growth factor receptor mutant–positive non–small-cell lung cancer. Recent evidence suggests that downregulation of gene of phosphatase and tensin homolog deleted on chromosome 10 plays an important role in acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitors in various types of cancers, including lung cancer. It was reported that the E3 ubiquitin ligase neural precursor cell expressed developmentally downregulated gene (NEDD4) (also known as NEDD4-1) negatively regulated phosphatase and tensin homolog deleted on chromosome 10 protein levels through poly-ubiquitination and proteolysis in carcinomas of the prostate, lung, and bladder. Whether this process plays a role in epidermal growth factor receptor-tyrosine kinase inhibitors resistance in non–small-cell lung cancer has not been studied extensively. In view of this, we investigated the involvement of NEDD4 and phosphatase and tensin homolog deleted on chromosome 10 in acquired erlotinib resistance with tyrosine kinase inhibitor–sensitive (HCC827) or tyrosine kinase inhibitor–resistant (Erlotinib-resistant HCC827/ER cells which harbored exon 19 deletion. Overexpression of NEDD4 in HCC827/ER cells was detected, and the reverse correlation between NEDD4 and phosphatase and tensin homolog deleted on chromosome 10 expression in these cells was also revealed. In HCC827/ER cells with knockdown of NEDD4, phosphatase and tensin homolog deleted on chromosome 10 and p-Akt expressions were decreased; the sensitivity of HCC827/ER cells to erlotinib was partially restored. Similar results were also observed in vivo. In H1650/ER cells harboring both exon 19 and phosphatase and tensin homolog deleted on chromosome 10 deletion, expression of p-Akt and sensitivity to erlotinib were not affected by simple knockdown of NEDD4 but affected after transfection of phosphatase and tensin homolog deleted on chromosome 10 into H1650/ER cells. Our results demonstrate that NEDD4 may promote the acquired resistance of non–small-cell lung cancer cells to erlotinib by decreasing phosphatase and tensin homolog deleted on chromosome 10 protein expression. Targeted decrease in NEDD4 expression may be a potential therapeutic strategy for tyrosine kinase inhibitor–resistant non–small-cell lung cancer.
Keywords
Introduction
Lung cancer is the most common cause of cancer-related death worldwide. 1 Non–small-cell lung cancer (NSCLC) is the most common type of lung cancer, with an overall 5-year survival rate of <20%. 2 Patients with activating mutation in epidermal growth factor receptor (EGFR) gene, exon 19 deletion, or L858R mutation occur in about 10%–15% of all NSCLC cases. 3 Tyrosine kinase inhibitors (TKIs), such as erlotinib and gefitinib, have been widely used to treat these patients, often with excellent therapeutic results. However, despite initial remission, recurrence was observed within 1 or 2 years (median: 12 months) by multiple mechanisms, such as EGFR T790M, MET (an oncogene) amplification, overexpression of hepatocyte growth factor (HGF) and decrease in gene of phosphatase and tensin homolog deleted on chromosome 10 (PTEN).4–8 Acquired resistance to EGFR-TKIs is usually inevitable. Therefore, explorations of the exact mechanisms and solutions to improve the sensitivity to TKIs are urgently required.
PTEN was originally identified as a tumor suppressor gene frequently lost from a region of chromosome 10q23 in a variety of human tumors. It is a central negative regulator of the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/Akt signaling cascade and influences multiple cellular functions, including cell growth, survival, proliferation, and migration in a context-dependent manner. 9 Existing evidence suggests that loss of PTEN is associated with TKI resistance, and downregulation of PTEN was identified in clinical specimens obtained from patients with acquired resistance to EGFR-TKIs. 10 Yamamoto et al. 11 proved that gefitinib-resistant (PC-9/GEF) cell lines showed a marked decrease in PTEN expression, and PTEN knockdown resulted in drug resistance to gefitinib in PC-9 cells. In our previous studies, low expression of PTEN messenger RNA (mRNA) and protein was detected in Erlotinib-resistant HCC827/ER cells, compared with its parental HCC827 cells. 12 Interestingly, the decrease in PTEN protein level was more significant, which indicates that protein post-translational modification may play an important role in the downregulation of PTEN protein.
Neural precursor cell expressed developmentally downregulated 4 (NEDD4, also known as NEDD4-1) is a prototypical protein in the family of E3 ubiquitin ligases. 13 Evidence shows that NEDD4 is a proto-oncogenic ubiquitin ligase for PTEN, which negatively regulates PTEN abundance via catalyzing poly-ubiquitination of PTEN and thus leads to proteolysis of PTEN protein in cells. 14 It has been reported that negative regulation of PTEN by NEDD4-mediated poly-ubiquitination is involved in several biological and pathological processes, such as axon branching, 15 keloid formation, 16 and hepatocellular carcinoma growth. 17 Inverse relationships between the expression of NEDD4 and PTEN have also been observed in human NSCLC. 18 However, recent work failed to find the relationship between NEDD4-mediated PTEN degradation and acquired resistance to EGFR-TKIs in NSCLC.
We focused on comparing the expression of NEDD4 and PTEN between HCC827 and HCC827/ER cells and identifying the possible mechanism underlying the acquired resistance to TKIs in NSCLC cells. A higher expression of NEDD4 and lower level of PTEN was detected in HCC827/ER cells. NEDD4 knockdown could partially restore the expression of PTEN protein and the sensitivity to erlotinib in HCC827/ER cells. In H1650/ER cells harboring both exon 19 and PTEN deletion, simple NEDD4 knockdown cannot strikingly increase the sensitivity to erlotinib unless transfecting PTEN into H1650/ER cells. Therefore, we hypothesized that PTEN degradation may play a bridging role in the NEDD4-induced acquired resistance to EGFR-TKIs in NSCLC.
Materials and methods
Cell lines, reagents, and groups
The human lung cancer HCC827 cell line was purchased from ATCC (ATCC® CRL-2868™). HCC827/ER cells were cultured by Dr Jing Han by applying high-dose (1–5 µM) pulses of erlotinib combined with continuous low-dose (0.01 µM) administration for >8 months (12). H1650/ER cells were presented by Dr Li Li (Department of Respiratory Disease, Daping Hospital, Third Military Medical University, Chongqing, China). The lung cancer cells were grown in RPMI-1640 medium containing 10% fetal bovine serum (Gibco BRL, Carlsbad, CA, USA) and 100 U/mL penicillin/streptomycin at 37°C in a humidified incubator containing 5% CO2. Erlotinib (OSI-744) was purchased from Selleck Chemicals (Houston, TX, USA).
Cell proliferation assay
Cells were spilt and plated into 96-well plates at a density of 4500 cells/well (in triplicate) for HCC827, HCC827/ER, HCC827/ER-EV, HCC827/ER-siNEDD4, H1650/ER-EV, H1650/ER-siNEDD4, H1650/ER-EV-PTEN, and H1650/ER-siNEDD4-PTEN cells. After adherent cell growth, the medium was replaced with complete medium containing increasing concentrations of 0, 0.001, 0.01, 0.1, 1, 10, or 100 µM erlotinib. After being cultured for 48 h, the viable cells were quantified using Cell Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan). Mixed liquor of 100 µL containing 10 µL CCK-8 solution and 90 µL medium was added to each well at the end of treatment. After being incubated for 1 h, the absorbance at 450 nm was measured using a spectrophotometer (Epoch; BioTek, Winooski, VT, USA). Each experiment was independently performed three times.
Lentivirus infection
To stably downregulate NEDD4 in HCC827/ER cells and express PTEN in H1650/ER cells, target cells were transfected with lentiviruses carrying siNEDD4, PTEN, or negative control vector (empty viral vector (EV)). These lentiviruses were packaged and purchased from Hanbio Biotechnology Co., Ltd (Shanghai, China). The infected cells were collected and the media were updated following cultivation for 12 h. The transfection efficiency was observed under a fluorescent microscope (BX50; Olympus, Tokyo, Japan).
Quantitative real-time polymerase chain reaction
Total RNA was extracted from harvested cells using RNAiso Reagent Plus (Takara Biotechnology, Dalian, China). Reverse transcription reactions were performed using an RT kit from Takara following the manufacturer’s protocol. The relative expression level of NEDD4 and PTEN mRNA was normalized to β-actin expression. The sequences of primers used in this experiment were as follows: NEDD4: forward: 5′-GGTGGAGGTGTTCGGGCT-3′, reverse: 5′-GCAAGGCCTATTCCGGCTA-3′; PTEN: forward: 5′-CGACGGGAAGACAAGTTCAT-3′, reverse: 5′-AGGTTTCCTCTGGTCCTGGT-3′; and β-actin: forward: 5′-GAGCTACGAGCTGCCTGACG-3′, reverse: 5′-CCTAGAAGCATTTGCGG TGG-3′. All reactions were performed in triplicate, and all experiments were conducted three times independently.
Western blotting
Harvested cells were washed twice with cold phosphate-buffered saline (PBS). The total protein was extracted using radioimmunoprecipitation assay (RIPA) buffer, denatured with loading buffer and separated by electrophoresis on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels, and then transferred onto polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). The membranes were blocked with 5% bovine serum albumin (BSA) (Wuhan Boster Bio-Engineering Co., Ltd, Wuhan, China) at room temperature for 1 h and subsequently incubated with the following antibodies: anti-NEDD4 (1:1000; Wuhan Proteintech Group, Inc., Wuhan, China), anti-PTEN (1:500; Wuhan Boster Bio-Engineering Co., Ltd), anti-Akt, anti-p-Akt (Ser473) (1:1000; Signalway Antibody, Boston, MA, USA), anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:1000; Wuhan Boster Bio-Engineering Co., Ltd) at 4°C overnight. After being washed three times with Tris-buffered saline containing Tween-20 (1:500), the membranes were incubated with secondary antibodies conjugated to horseradish peroxidase (Wuhan Boster Bio-Engineering Co., Ltd) for 1 h at room temperature. Protein bands were visualized using an enhanced chemiluminescence detection kit (Pierce Biotechnology, Inc., Rockford, IL, USA). Band density was quantified with Quantity One software (Bio-Rad Laboratories, Inc., Hercules, CA, USA). GAPDH was used as an internal control.
In vivo experiments
Nude mice (female, 4–6 weeks old) were purchased from the Chinese Academy of Medical Sciences (Beijing, China) and housed in groups (three mice per cage) in a laminar flow room under specific pathogen-free conditions at room temperature (22 ± 2°C) and humidity (<40%), all the food and water were free to access. A total of 12 mice were randomized into four groups (three mice per group) in this study. A total of 2 × 106 HCC827, HCC827/ER, siNEDD4-, or EV-infected HCC827/ER cells were subcutaneously injected into the mouse skin under the left lower quadrants, respectively. When tumor volumes reached about 100–150 mm3, these mice were treated with intragastric administration of erlotinib (60 mg/kg) for 2 weeks once a day. Tumor sizes were measured every 3 days.
Statistical analysis
Date analysis was performed using SPSS 20.0 software. Quantitative data were presented as mean ± standard deviation (SD). Group comparison was performed by Student’s t test. p value < 0.05 was considered statistically significant.
Results
Both NEDD4 mRNA and protein were overexpressed in TKI-resistant cells compared with TKI-sensitive cells
The CCK-8 assay revealed the sensitivity of the HCC827 and HCC827/ER cells to erlotinib. HCC827/ER was clearly more resistant to erlotinib than HCC827 (Figure 1(a)). To determine the role of NEDD4 in acquired resistance to EGFR-TKIs in NSCLC, we analyzed the expression of NEDD4 mRNA and protein in HCC827 and HCC827/ER cells (Figure 1(b) and (c)). Quantitative RT-PCR (q-PCR) showed that the NEDD4 mRNA level in HCC827/ER cells was higher than that in HCC827 cells (Figure 1(b)). Subsequent western blot analysis demonstrated that NEDD4 expression was also significantly increased in HCC827/ER cells (Figure 1(c)). All these suggest that the overexpression of NEDD4 may contribute to secondary resistance in HCC827/ER cells.

NEDD4 is overexpressed in HCC827/ER cells and has an inverse relationship with PTEN. (a) The sensitivity to erlotinib of HCC827 and HCC827/ER cells was determined by CCK-8 assay. IC50 of groups HCC827 (0.10 ± 0.02 µM), HCC827/ER (11.6 ± 0.90 µM) (n = 3; *p < 0.05). (b) The mRNA expression of NEDD4 and PTEN was analyzed by RT-qPCR. (c) The protein level of NEDD4, PTEN, Akt, and p-Akt was detected by western blots. (d) HCC827/ER cells were treated with 1 µM MG132 for 12 h; the PTEN level was partially restored.
NEDD4 promotes TKI resistance through Ub-mediated degradation of PTEN protein in NSCLC cells
In this study, we detected PTEN expression at mRNA and protein levels by q-PCR and western blot, respectively, and found that PTEN was decreased at both levels in TKI-resistant cells (Figure 1(b) and (c)). The PI3K/Akt signaling, a downstream of PTEN, was activated (Figure 1(c)). More interestingly, the decrease in PTEN mRNA level was about 50%, and in PTEN protein level about 85%, obviously higher than the former. The PTEN protein level was negatively correlated with NEDD4, and the decrease in PTEN could be restored by MG132, a proteasome inhibitor (Figure 1(d)).
NEDD4 depletion enhanced the sensitivity of HCC827/ER to erlotinib by upregulating PTEN protein levels
To evaluate the role of NEDD4 in acquired erlotinib resistance in HCC827/ER cells, we knocked down the expression of NEDD4 with lentivirus-carrying siNEDD4 (Figure 2(a)) and checked the sensitivity changes under erlotinib treatment (Figure 2(b)). We verified that the depletion of NEDD4 resulted in upregulation of PTEN protein and inhibited the activation of PI3K/Akt signaling (Figure 2(c)). Moreover, the depletion of NEDD4 led to a significant decrease in IC50 value compared with the control group. These data indicated that NEDD4 depletion enhanced the sensitivity of lung cancer cells to erlotinib.
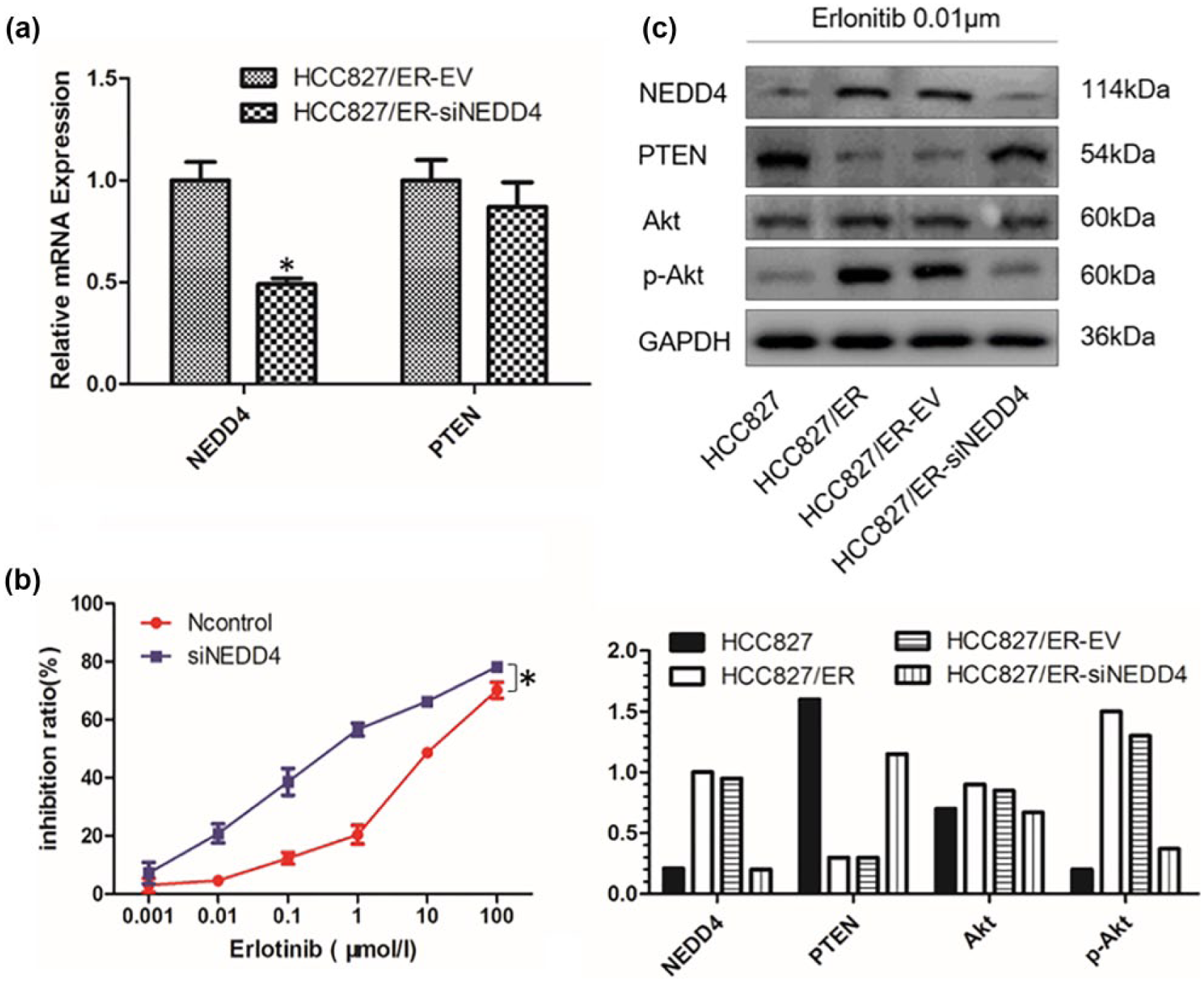
NEDD4 knockdown enhanced cell sensitivity to erlotinib by upregulating the expression of PTEN protein but not mRNA. (a) The expression of NEDD4 was knocked down in HCC827/ER cells; the expression of NEDD4 and PTEN mRNA was analyzed by RT-qPCR (n = 3; *p < 0.05). (b) The sensitivity of HCC827/ER-EV and HCC827/ER-siNEDD4 cells to erlotinib was determined by CCK-8 assay. IC50 of HCC827/ER-EV (13.88 ± 1.96 µM), HCC827/ER-siNEDD4 (0.72 ± 0.12 µM) (n = 3; *p < 0.05). (c) The protein level of NEDD4, PTEN, Akt, and p-Akt was detected.
Decreased expression of NEDD4 enhanced the sensitivity to erlotinib and increased the treatment benefit from erlotinib in nude mice
In this study, we further confirmed the above findings by establishing an in vivo model. After the tumor mass was palpable in all groups, the size of these tumors was measured, and the growth curves were plotted using GraphPad Prism 5 (Figure 3(b)). All mice were sacrificed 2 weeks after treatment and the tumor mass was measured (Figure 3(a)). The tumor volume of the HCC827/ER-injected mice was approximately five times larger than that of the HCC827 group. Inhibition of tumor growth was observed in siNEDD4-bearing nude mice. The tumor mass in the siNEDD4 group achieved 30% of that in the control (p < 0.05).

Decrease in NEDD4 increased the treatment benefit from erlotinib in nude mice. (a) Representative nude mice bearing xenografts in four groups. All the mice received erlotinib through intragastric administration for 15 days. The mice were sacrificed and the tumor was measured. (b) Growth curves of tumors in four groups (n = 3;*p < 0.05). (C) The weight of these tumors (n = 3;*p < 0.05).
NEDD4 increased NSCLC cell resistance to erlotinib through PTEN degradation
To verify the function of PTEN in the acquired erlotinib resistance in NSCLC cells, a repeated experiment was performed in H1650/ER cells, which harbor both exon 19 and PTEN deletion. Compared with cells in the control group, NEDD4 downregulation or PTEN transfection alone cannot inhibit the resistance to erlotinib in H1650/ER cells (Figure 4(b)). However, in the group of combined transfection of siNEDD4 and PTEN, the expression of PTEN protein was higher than that in the group of single PTEN transfection, and the sensitivity to erlotinib in H1650/ER cells was significantly increased (Figure 4(b) and (c)). These findings suggest that the PTEN degradation induced by NEDD4 plays a key role in the acquired resistance to EGFR-TKIs in NSCLC.

Only co-transfection of siNEDD4 and PTEN can restore the sensitivity to erlotinib in H1650/ER cells. (a) Contrasting the expression of NEDD4 in H1650 cells transfected with siNEDD4 or EV. (b) Determining the sensitivity of H1650/ER-EV, H1650/ER-siNEDD4, H1650/ER-PTEN, and H1650/ER-siNEDD4-PTEN cells to erlotinib. IC50 of each group: H1650/ER-EV (47.33 ± 5.54 µM), H1650/ER-siNEDD4 (28.31 ± 2.37 µM), H1650/ER-PTEN (29.75 ± 3.73 µM), H1650/ER-siNEDD4-PTEN (4.64 ± 1.17 µM) (n = 3;*p < 0.05). (c) The expression of NEDD4 and PTEN in four groups.
Discussion
PI3K/Akt signaling pathway is associated with cell proliferation survival. The disturbed activation of PI3K/Akt signaling pathway plays a major role not only in tumor growth but also in the response of a tumor to treatment. Evidence showed that the inhibition of PI3K/Akt pathway could inhibit the proliferation and induce the apoptosis of cancer cells. 19 PI3K inhibitors also markedly enhanced the sensitivity of NSCLC cells with high level of phosphorylated Akt to drug-induced apoptosis.20,21 Blocking the PI3K/Akt signaling pathway can overcome gefitinib resistance in NSCLC cell lines. 22 In our previous studies,12,23 we confirmed that PI3K/Akt signaling pathway was activated in either PC9/ER or HCC827/ER cells. In PC9/ER cells, we demonstrated that the decrease in miR-223 induced the acquired resistance to EGFR-TKIs by activating the IGF1R/PI3K/Akt pathway but failed to find the decrease in PTEN. However, the downregulation of PTEN protein, a central negative regulator of PI3K/Akt pathway, was found in HCC827/ER cells, which indicated that the decrease in PTEN may play a key role in the activation of PI3K/Akt pathway and the resistance to TKIs in HCC827/ER cells.
PTEN is a dual-specificity protein and lipid phosphatase. In terms of function, it regulates many cellular processes, including proliferation, survival, energy metabolism, cellular architecture, and motility.24–28 However, it is more widely known as a tumor suppressor gene and its tumor-suppressing activity largely depends on its lipid phosphatase activity which opposes PI3K/Akt activation. 29 The link between PTEN and PI3K/Akt pathways was confirmed by the finding that loss of PTEN function would enhance the activation of Akt via increasing the intracellular levels of phosphatidylinositol (3, 4, 5) trisphosphate (PIP3) in cells. 30 Moreover, it has been demonstrated that subtle changes in the dose of PTEN can affect the activation of Akt and have profound effects on tumor susceptibility.31,32 Thus, regulating the expression of PTEN may have a profound influence on tumor treatment.
Recently, NEDD4 has been identified as a proto-oncogenic ubiquitin ligase for PTEN. 14 Previous studies have confirmed that NEDD4 is involved in a variety of physiological processes of tumor with or without the PI3K/PTEN/Akt pathway. For example, NEDD4 is overexpressed in colorectal cancers and promotes growth of colon cancer cells independently of PTEN and PI3K/Akt signaling. 33 Fouladkou et al. 34 failed to support the role of NEDD4 as the E3 ligase in regulating PTEN stability and subcellular localization. But the dominant views are still that the oncogenic activity of NEDD4 depends on the degradation of PTEN. Amodio et al. 18 have demonstrated that NEDD4 is overexpressed in human NSCLC, and the overexpression of NEDD4 leads to an increase of the ubiquitylated form of PTEN, thereby reducing PTEN levels and increasing Akt activation. Ahn et al. 35 also confirmed that NEDD4 can inhibit PTEN-induced apoptosis in cells; in addition, PTEN can act as a feedback regulator for NEDD4 and transcriptionally regulate the expression of NEDD4. However, the role of NEDD4 in resistance to EGFR-TKIs is rarely reported.
In this study, we assessed the expression levels of NEDD4 and PTEN in HCC827/ER cells and found that the expression of NEDD4 mRNA was twice greater than that in HCC827 cells. Western blot analysis also demonstrated an increase in the levels of NEDD4 protein in HCC827/ER cells. On the contrary, PTEN mRNA expression was reduced by 50% in HCC827/ER cells. However, the decrease in PTEN protein was about 85%, which is more significant than that in the levels of PTEN mRNA. These results revealed an inverse relationship between the expression of NEDD4 and PTEN in HCC827/ER cells, indicating that NEDD4-mediated PTEN downregulation may play an important role in the resistance to erlotinib in HCC827/ER cells. Whereupon, we knocked down the expression of NEDD4 in HCC827/ER cells and found that the PI3K/PTEN/Akt pathway was inhibited and the resistance to erlotinib was partially reversed. Similar results were found in an in vivo study. These findings confirmed that the overexpression of NEDD4 activates the PI3K/Akt pathway by reducing PTEN abundance in HCC827/ER cells and induces resistance to erlotinib. To clarify the role of PTEN in the resistance induced by NEDD4, we knocked down the expression of NEDD4 in H1650/ER cells. However, no striking increase in the sensitivity to erlotinib was found. Then, we transfected PTEN into H1650/ER cells. A significant increase in the sensitivity to erlotinib was found in the group of combined transfection of siNEDD4 and PTEN, which suggests that the NEDD4-induced NSCLC resistance to erlotinib depends on the PTEN degradation.
Therefore, NEDD4 could be a potential indicator for NSCLC patients with EGFR-TKI resistance, and targeting NEDD4 might be a promising strategy for enhancing the sensitivity to EGFR-TKIs in the patients without PTEN deletion.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported in part by grants from the National Natural Science Foundation of China (81672841) and the National High Technology Research and Development Program of Chongqing, China (2011AB5032).