
Abstract
Imatinib mesylate, a BCR/ABL fusion protein inhibitor, is the first-line treatment against chronic myelogenous leukemia. In spite of its advantageous viewpoints, imatinib still has genuine impediments like undesirable side effects and tumor resistance during chemotherapy. Nanoparticles with sustainable release profile will help in targeted delivery of anticancer drugs while minimizing deleterious side effects and drug resistance. The use of biopolymers like galactoxyloglucan (PST001) for the fabrication of imatinib mesylate nanoparticles could impart its use in overcoming multidrug resistance in chronic myelogenous leukemia patients with minimal side effects. This study involved in the synthesis of PST-Imatinib nanoconjugates with appreciable drug payload and excellent cytotoxicity against drug-resistant chronic myelogenous leukemia cell line (K562) in comparison with free drug. The use of bioinformatics tool revealed better binding affinity for the drug–polysaccharide complex than the drug alone with three proteins: 3QX3 (Topoisomerase), 1M17 (EGFR tyrosine kinase domain), and 3QRJ (ABL1 kinase domain). Assessment of the biochemical, hematological, and histopathological parameters in mice upheld the security and adequacy of the nanoconjugate compared to free drug. Although perspective investigations are warranted, in a condition like drug resistance in leukemia, this nanoconjugate would display a productive approach in cancer therapeutics.
Introduction
Targeted therapy embattling active tyrosine kinases in chronic myelogenous leukemia (CML) has improved treatment outcome significantly. BCR/ABL is often regarded as the molecular signature in CML, and the use of imatinib mesylate has transformed CML management approach. Compared to the previously accepted therapy combining interferon and cytarabine, imatinib gives better response, prognosis, and fewer side effects. Imatinib executes its activity by competitively inhibiting the ATP binding site of BCR/ABL oncoprotein which results in the apoptosis of Ph+ hematopoietic cells sparing the normal cells. 1 In spite of its highly specific activity, 33% of CML cases do not achieve complete cytogenetic response either due to drug resistance or intolerable drug-related complications. 2 Imatinib treatment failure is often managed by dose escalation but is likely to be effective only in a subset of patients with previous cytogenetic response. Resistance to imatinib has prompted research in alternate drug resistance mechanisms out of which drug transporter status is the mainstream target. Development of drug resistance is often considered a multifactorial phenomenon in which overexpressed drug efflux transporter is an imperative contributor. In addition, modulation of P-glycoprotein (Pgp) expression during imatinib therapy could drastically reduce drug dosage. 3 More frequent observation of Pgp overexpression in advanced stage of CML 4 and reduction in critical concentration needed for anti-leukemic effect associated with intramembranous drug transporters 5 emphasized the need to explore the link between Pgp expression and imatinib resistance.
Numerous studies in the course of recent years have demonstrated that nanoparticle formulations possess ability to make drug-resistant tumors vulnerable to chemotherapy.6–8 Targeted drug delivery systems using nanoparticles help in the delivery of payload to tumor cells by overcoming drug resistance. The use of biocompatible biopolymers in the fabrication of imatinib mesylate nanoparticles could help in overcoming multidrug resistance in CML patients. Hence, the present investigation was aimed to synthesize and evaluate the cytotoxic efficiency of imatinib mesylate nanoparticles using natural galactoxyloglucan for BCR/ABL specific leukemia therapy.
Methodology
Materials
Tissue culture reagents including Dulbecco’s Modified Eagle Medium (DMEM) and antibiotic–antimycotic mixture were purchased from Invitrogen (Carlsbad, CA) supplemented with fetal bovine serum (FBS; Invitrogen, CA, USA). All chemicals were purchased from Merck Chemicals, India.
Isolation of polysaccharide PST001 from tamarind seed kernel
Polysaccharide (PST001) was isolated from seed kernel of Tamarindus indica as reported earlier. 9 Briefly, the powdered material was treated with petroleum ether for 72 h to remove any fat present, followed by hot water extraction using Soxhlet apparatus. The filtrate was subjected to ethanol precipitation at 4°C, followed by dialysis against double distilled water using 12,000–14,000 MW membrane (Spectra/Por, MWCO 12,000–14,000 Da; Spectrum Laboratories, CA, USA). The solution was treated with chloroform to remove protein, if present, in a separation funnel until the interphase became clear. The steps were repeated several times until the water chloroform interphase became clear. The aqueous phase was again dialyzed, concentrated, and precipitated with ethanol. Polysaccharide precipitate was collected by centrifugation at 3000g for 20 min at 4°C, re-dissolved in distilled water, and lyophilized. The isolated polysaccharide was purified with high performance liquid chromatography (Shimadzu, Japan) using GPC/SEC columns (Agilent Technologies, USA). 10
Preparation and characterization of nanoparticles
Preparation
The imatinib mesylate encapsulated polysaccharide nanoparticles were prepared by dimethyl sulfoxide (DMSO) method. Briefly, 100 mg PST001 and imatinib mesylate (5 mg) were dissolved in 5 mL of DMSO. This solution was added drop wise to 20 mL aqueous solution containing 1.5% PVA (polyvinyl alcohol) and homogenized at 18,000g. After homogenization, the nano-emulsion was stirred for 5 h under dark at room temperature. The resulting mixture was centrifuged at 20,000g for 30 min to pellet down the nanoparticles. The pellet was washed and kept for dialysis with several water changes for 4 days to remove free drug and excess DMSO. Then, the nanoparticles were freeze-dried and stored at 4°C until further use.
Particle size, shape, encapsulation efficiency, and drug content
PST-Imatinib (PST-IM) nanoparticles were initially characterized by transmission electron microscopy (Jeol/JEM 2100). Particle size distribution, polydispersity index, and zeta potential of PST-IM nanoparticles were determined by dynamic light scattering (DLS) with the help of a nanoparticle analyzer SZ-100 (Horiba, Japan) at scattering angle of 90°. The molecular arrangement and vibrations arose during the formation of PST-IM nanoparticles were studied by Fourier Transform Infrared Spectroscopy (FTIR). Briefly, 2 mg lyophilized PST-IM nanoparticles were analyzed using FTIR (Thermo Nicolet, Avatar 370) with a spectral range of 4000–400 cm−1.
The loading content of imatinib mesylate in PST-IM nanoparticles was measured by ultraviolet (UV)-visible spectrophotometric assay. Briefly, 10 mg nanoparticles were dissolved in 2 mL phosphate-buffered saline (PBS) and absorbance was read at 265 nm in a UV-visible spectrophotometer (Bio Spec-1601, Shimadzu, Japan) to determine the drug content from a standard graph.6,7 The percentage of encapsulation efficiency (EE%) and drug content (DC%) of PST-IM nanoparticles were determined using the following two formulas

The in vitro drug release of PST-IM nanoparticles was carried out using the previously described method with some modifications. Briefly, 10 mg of nanoparticles was suspended in 2 mL PBS and transferred into a dialysis bag. The dialysis bag was then placed into a 100 mL bottle containing 50 mL PBS (pH 7.4) as well as citrate buffer (pH 5.8) and was stirred at 2000g at 37°C. While stirring, 1 mL sample was withdrawn at different time points from the bottle for 10 days. To maintain the volume, 1 mL fresh buffer was added to the bottle after each withdrawal. Drug content of each sample was measured spectrophotometrically at 265 nm with buffer as blank. 7
In silico analysis of PST-IM nanoparticles
Tools used for in silico studies
We have optimized the model of PST-IM nanoparticles at M062X level of density functional theory. Optimized geometry was confirmed to be a minimum by computing the vibrational frequencies. All geometry optimizations were done using Gaussian 09 suite of computational chemistry software package. 11 The output was analyzed using Chemcraft 12 visualization software. The optimized geometries were docked against various proteins downloaded from RSC using Glide of Schrodinger Software suite.13–15
Target identification
The three dimensional structure of the target proteins human topoisomerase II beta in complex with DNA and etoposide (PDBID–3QX3), epidermal growth factor receptor complexed with erlotinib (PDBID–1M17), and human ABL1 kinase (PDBID–3QRJ) are the targets which were selected for this study and were downloaded from protein structure repository protein data bank. The structure of the proteins was refined using the Schrodinger software suite, and the active site was identified using sitemap module of the same software which predicts the binding site in three stages.
Preparation of lead compound
The three dimensional structure of imatinib was retrieved from PubChem compound database and was subjected for optimization to get a stable structure using Gaussian 09 (G09) suite software of computational chemistry programs. The energy-minimized structure of the compound is needed for docking studies. Geometry optimization of a compound is one of the significant steps in drug design that attempts to find the configuration of minimum energy, that is, calculation to find a more stable molecule. The lead optimization can alter the structure of lead candidates to improve properties in order to make them more effective and safer. Docking procedures aim to identify accurate poses of ligands in the binding pocket of a protein and to predict the affinity between protein and the ligand. All the docking analyses were conducted with the aid of “grid-based ligand docking with energetics” (Glide) tool included in the Maestro 9.3 (Schrodinger) software. The best pose was selected based on the Glide score.
In vitro analysis of PST-IM nanoparticles
Drug-resistant cell line development and characterization
Ph-positive CML cell line K562 was obtained from NCCS (Pune, India) and cultured in RPMI 1640 (Sigma-Aldrich, USA) supplemented with 10% FBS (Invitrogen, USA). The cells were incubated at 37°C under normoxic conditions and 5% CO2. CML cells were maintained in exponential growth phase and resistance developed by treating the cells with gradual stepwise increasing concentrations of imatinib starting from 2 nM. Drug concentration was increased at every passage at a rate of 0.2 nM till 1 µM resistance was achieved and designated as resistant K562 cell line (K562R). The resistance of K562 cell line was characterized previously by Gene knock down experiment followed by western blotting. 16 Jurkat cell line and HL-60 cell lines purchased from NCCS (Pune, India) were used as Ph-negative cell lines for the study.
In vitro cytotoxicity assay
The growth inhibitory capacity of the PST-IM nanoparticles was evaluated in the above-mentioned panel of cancer cell lines by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium) assay as previously reported.9,17 In brief, 5000 cells/well were seeded in 96-well plates. The cells were treated with varying concentration of drugs for 24, 48, and 72 h of incubation. The cells were then harvested by microplate centrifugation. MTT dye was added, and the formazan crystals were dissolved using lysis buffer. The absorbance was measured at 570 nm using a microplate spectrophotometer (BioTek, Power Wave XS). The assay was done in triplicates, and the proliferation rate and inhibitory rate of the cells were calculated with the following formulas

MTT assays were performed on K562, K562R, Jurkat, and HL-60 with various concentrations of PST-IM nanoparticles over a period of 24–72 h.
Apoptosis evaluation
Morphological assessment of apoptosis was conducted in cells treated with PST-IM nanoparticles (1 µg/mL) using the acridine orange–ethidium bromide dual staining procedure. These experiments were performed as described earlier, 9 and the cells were observed under an inverted fluorescent microscope (Olympus 1X51, Singapore), using an FITC filter. The nuclear condensation studies in cells treated with nanoparticles were analyzed using Hoechst staining procedure. 9 The cells were observed under an inverted fluorescent microscope, using a 4′,6-diamidino-2-phenylindole (DAPI) filter to view the nuclear condensation of treated cells compared to control.
RNA isolation and complementary DNA synthesis
Total RNA was isolated using TRIzol Reagent (Sigma-Aldrich, St Louis, MO) from K562 and K562R treated with PST-IM nanoparticles. RNA cleanup and DNase digestion were performed using Amplification Grade DNase I kit (Sigma-Aldrich, St Louis, MO). RNA was checked for integrity and purity by gel electrophoresis and UV–visible spectroscopy (Bio Spec-1601, Shimadzu, Japan). RNA concentration and A260/A280 ratios were also calculated. RNA integrity number (RIN) values were calculated using Agilent Bioanalyzer 2100 (Agilent Technologies, PaloAlto, CA, USA), and only the samples with A260/A280P1.8 and RIN values above 6 were used for further experiments. High Capacity cDNA Reverse Transcription Kit was used as per the manufacturer’s instruction (Applied Biosystems, USA) for complementary DNA (cDNA) preparation.
Gene expression analysis
Diluted cDNA was added to 1× polymerase chain reaction (PCR) master mix (Orion Diagnostics, India) and 0.5 µM each of forward and reverse primers (Table 1). The reaction consisted of an initial activation of polymerase at 95°C for 15 min followed by 32 cycles of denaturation at 95°C for 30 s, annealing at 58°C for 45 s, extension at 72°C for 45 s, followed by a final extension at 72°C for 5 min. The PCR product was analyzed using 2% agarose gel electrophoresis followed by detection of bands using Gel Doc (BioRad, USA).
Details of the primer used.

Western blot analysis
Proteins were extracted from K562 and K562R cells treated with PST-IM nanoparticles using radioimmunoprecipitation assay (RIPA) buffer (Thermo Scientific, IL, USA). Protein concentrations were determined using Coomassie plus protein assay reagent and bovine serum albumin (BSA) standards (Pierce, IL, USA). Approximately, 20 µg of proteins was separated on 10% SDS-PAGE and transferred to polyvinylidene-difluoride membrane (Millipore, MA, USA). The membrane was blocked using 5% of BSA (Santa Cruz Biotechnology Inc., TX, USA) and incubated with the specific primary antibodies ABCB1 (Cell Signaling, USA) and β-actin (Sigma-Aldrich, MO, USA). Horseradish peroxidase (HRP)–conjugated rabbit anti-mouse IgG secondary antibody (Cell Signaling) was used against the primary antibodies and stained with ECL detection kit (BioRad, USA) and image analyzed in Flourchem (ProteinSimple, USA).
In vivo toxicity analysis
Animals and treatments
Subacute toxicity of PST-IM nanoparticles was evaluated in BALB/C mice weighing about 25–30 g. Animals were divided into three groups (group 1, group 2, and group 3), each group containing six adult females. Group 1 received 2 mL PBS and was treated as a control. Group 2 received PST-IM nanoparticles (50 mg/kg imatinib mesylate in nanoparticles) suspended in PBS. Group 3 received 50 mg/kg free drug (imatinib mesylate in PBS). Treatment for each group was done per orally (PO) using oral gavage for 28 days. 7 Throughout the study period, animals were monitored for the development of any toxicological signs and symptoms such as abnormal posture, abnormal movements, difficulties in respiration, and changes in body weight and feed intake. To know the potential side effects at the cellular level, blood biochemistry and hematological parameters were examined in treated groups of animals and compared with the control group. On the 29th day, the animals were sacrificed by cervical dislocation; the blood, serum, and internal organs were collected and used for further hematological, biochemical, and histopathological evaluation. Animal experimentation protocols were reviewed and approved by the Institutional Animal Ethics Committee (IAEC) of the Regional Cancer Centre, Trivandrum, India.
Histopathological evaluation
The internal organs were fixed in 10% phosphate-buffered formalin for 24 h. Samples were routinely processed and embedded in paraffin, and 4 µm sections were de-paraffinized in xylene and then rehydrated in absolute ethanol. The sections were then rinsed with 95% alcohol followed by 70% alcohol for 2 min each. They were washed in distilled water and stained with Mayer hematoxylin solution for 8 min. The sections were washed under warm running tap water and rinsed in distilled water. Ten dips of 95% alcohol was given and then counterstained with eosin-phloxine B for 1 min. The sections were then dehydrated through 95% alcohol followed by two changes of absolute alcohol. The sections were cleared in xylene, dried, and mounted on slide. The dried slides were observed under a microscope (Olympus 1X51, Singapore).
Results
Characterization of nanoparticles
Galactoxyloglucan (PST001) was isolated from seed kernel of T. indica and purified by high performance liquid chromatography. We have synthesized nanoencapsulated imatinib mesylate within PST001. Previous studies proved that the co-encapsulation of doxorubicin with PST001 exhibited minimal cardiotoxicity and significant tumor specific cytotoxicity. 18
Particle size of nanoparticles is an important parameter determining the biological activity and the efficiency of cellular uptake. PST-IM nanoparticles were found to have an average diameter of 206 nm with polydispersity index of 0.5. The particle size determined by DLS is shown in Figure 1. Zeta potential is another factor determining the stability of nanoparticles in aqueous medium as the surface charge controls the aggregation of these nanoparticles. The negative zeta potential (−29.3 mV) indicates the absence of aggregation of nanoparticles (Figure 2).

Particle size distribution—DLS spectra of PST-IM nanoparticles.

Zeta potential of PST-IM nanoparticles.
PST001 undergoes gelation in DMSO–water solvent system followed by imatinib mesylate encapsulation. The size of the nanoparticle, which was evaluated using transmission electron microscope (TEM) at an accelerated voltage of 200 kV, clearly showed that the majority of the particles were uniform with an average size of 50 ± 5 nm (Figure 3(a)). The Selected Area Electron Diffraction (SAED) pattern of nanoparticles is shown in Figure 3(b).

(a) TEM image and (b) SAED pattern of PST-IM nanoparticles.
The aromatic groups in the imatinib molecule were expected to interact with PST001 during the formation of the PST-IM nanoparticle. In the FTIR spectrum, the peak change in 3650–3050 cm−1 due to the extensive intra-molecular hydrogen bonding indicated that the intra-molecular interactions were broadened, leading to the intermolecular interactions between the PST001 and imatinib mesylate. The characteristic peaks of imatinib observed includes 1475, 1607, 2922, and 3334 which are assigned to the stretching vibration of aromatic C=C, amide (mainly C=O stretching vibrations), C–H stretch, and O–H stretch, respectively, extend in the PST-IM conjugate, reinforcing the inter-species interactions that have resulted at the time of formation of the PST-IM conjugate. There could be physical interactions between the functional groups of the imatinib and polysaccharide by the formation of intermolecular hydrogen bonds with functional groups of PST001 and imatinib mesylate in addition to the intermolecular interactions observed, as shown in Figure 4.
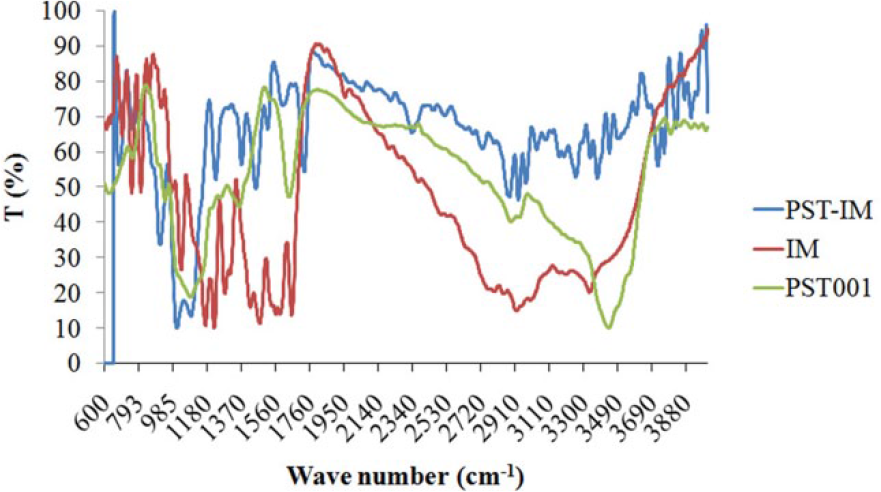
FTIR spectrum of PST-IM nanoparticles.
Encapsulation efficiency, drug load, and in vitro drug release kinetics
The nanoparticles showed an encapsulation efficiency up to 60% when evaluated using UV-visible spectrophotometer with a drug payload of 3.12% (0.0312 mg/mg nanoparticles). As graphically illustrated in Figure 5, in vitro drug release was found to be approximately 63% of total encapsulated imatinib mesylate released from nanoparticles during the first 24 h of incubation at pH 7.4. After the initial burst release, imatinib mesylate was released continuously at a linear rate with respect to the square root of time, reflecting diffusion-mediated release from the nanoparticles. The polysaccharide enables an enormous water penetration causing the rapid drug release kinetics during initial 3 days followed by steady release for further 5 days in aqueous buffer.
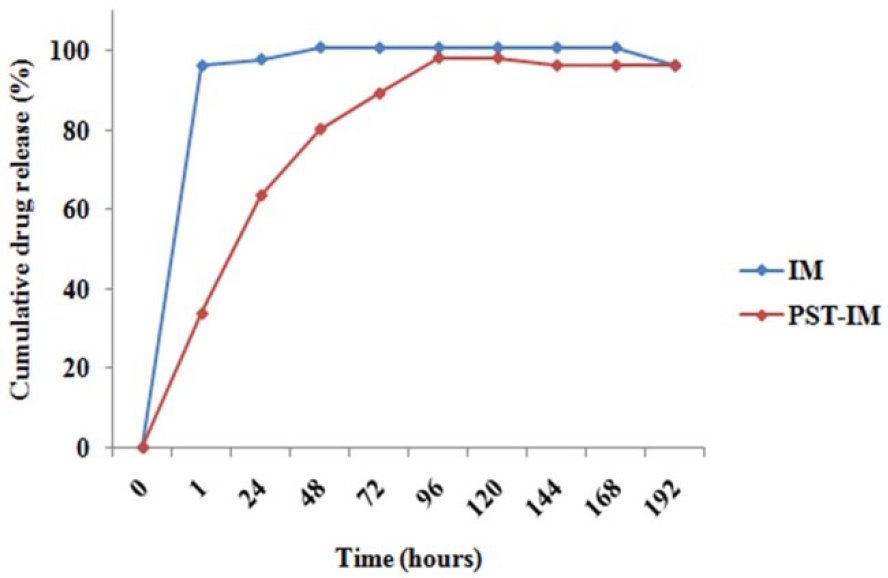
In vitro drug release kinetics of imatinib mesylate from nanoparticles compared to free drug at pH 7.4.
In silico analysis of PST-IM nanoparticles—active site identification, molecular docking of lead compound
Schrodinger software suite was used to find out the active site of the drug where the polysaccharide can be attached. Energy minimized structure of drug “Imatinib” was with Human Oxy hemoglobin (Hb) protein. The binding interaction shows the affinity of −5.0 and Glide score value −5.1.
Docking analysis was performed using a Glide tool for finding the interaction between the imatinib as well as PST-IM complex with 3QX3 (Topoisomerase), 1M17 (EGFR tyrosine kinase domain), and 3QRJ (ABL1 kinase domain). Docking simulations of imatinib and PST-IM complex resulted in better interaction with 3QX3. When imatinib was docked into the target protein, 3QX3 has hydrogen bonding interaction with the amino acid of DC8 and DT9, as depicted in Figure 6. In case of PST-IM complex, the result obtained as the hydrogen bond interaction of imatinib with a DNA base pair of DT9 protein according to glide/dock score, as depicted in Table 2. The DNA base pair such as DC11, DG10 and amino acids ARG820 and SER818 of the proteins has hydrogen bond interaction with the model.

Binding interaction of (a) imatinib with 3QX3 (Topoisomerase) and (b) PST-IM complex with 3QX3 indicating better interaction of drug–polysaccharide than drug alone. The 2D workspace of (c) Imatinib and (d) PST-IM complex with its interacting residues of 3QX3 (Topoisomerase).
Binding interaction of drug with 3QX3 (Topoisomerase).

Elec: electrostatic potential.
The imatinib bound to the target shows hydrogen bonding interaction with the amino acid of MET769 protein. When PST-IM complex docked with the target EGFR tyrosine kinase domain, the result obtained as the hydrogen bond interaction of imatinib with the amino acid of ASP831 and MET769 based on glide/dock score, as in Table 3. GLU780 residue of 1M17 (EGFR tyrosine kinase domain) has an hydrogen bond interaction with the model, as depicted in Figure 7.
Binding interaction of drug with 1M17 (EGFR tyrosine kinase domain).

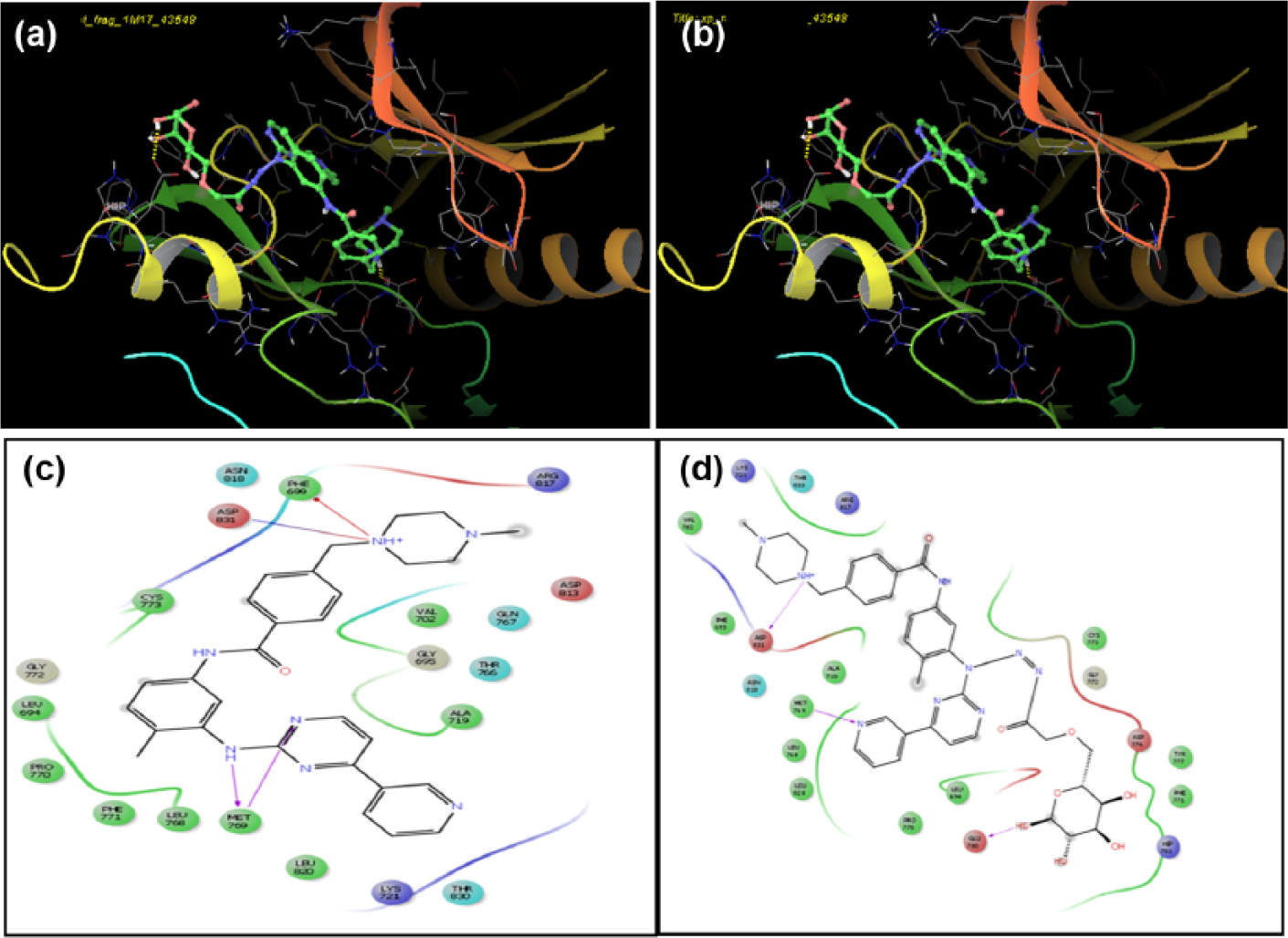
Binding interaction of (a) imatinib with 1M17 (EGFR tyrosine kinase domain) and (b) PST-IM complex with 1M17 indicating better interaction of drug–polysaccharide than drug alone. The 2D workspace of (c) imatinib and (d) PST-IM complex with its interacting residues of 1M17 (EGFR tyrosine kinase domain).
When imatinib was docked into the target protein 3QRJ (BCR/ABL), the hydrogen bonding interaction with the amino acid of ASP381, ILE360, and GLU282 protein has the binding affinity of −5.3 and Glide Score value of −5.8. The PST-IM complex was docked with the target and it was seen that the amino acid ASP381 had hydrogen bond interaction with the drug whereas the remaining residues such as ARG362, ARG386, SER 385 and LYS400 of 3QRJ showed hydrogen bond interaction with the model, and the drug PST001 complex has the binding affinity of –9.5 and glide score of –9.8. This shows better interaction than the drug, as shown in Table 4 and Figure 8.
Binding interaction of drug with 3QRJ (BCR/ABL).


Binding interaction of (a) imatinib with 3QRJ (BCR/ABL oncoprotein) and (b) PST-IM complex with BCR/ABL oncoprotein indicating better interaction of drug–polysaccharide than drug alone. The 2D workspace of (c) imatinib and (d) PST-IM complex with its interacting residues of 3QRJ (BCR/ABL oncoprotein).
In order to confirm whether the action of drug is via polysaccharide interaction with amino acid residues of active site or by drug interaction, polysaccharide alone was allowed to dock with the topoisomerase and EGFR tyrosine kinase domain. The model bound to the target 3QX3 and the result showing a dock score and glide score of –10.7 suggests the high interaction of PST001 with amino acid residues of 3Qx3. The 1M17 (EGFR tyrosine kinase domain) got the dock and glide score of −10.5 when docked with the model. Finally, one more docking has done with 3QRJ—the results form the docking score of −8.6 and glide score value of −8.6. From the results, we can understand that when we docked the PST-IM complex as well as the model of polysaccharide against three different proteins, highest binding interaction was observed for the interaction with DNA topoisomerase 3QX3, as depicted in Figure 9.

Binding interaction of (a and b) PST001 with 3QX3 (Topoisomerase), (c and d) PST001 with 1M17 (EGFR tyrosine kinase domain), and (e and f) PST001 with 1M17 (EGFR tyrosine kinase domain) indicating better interaction of polysaccharide with 3QRJ (BCR/ABL oncoprotein).
Absorption, distribution, metabolism and excretion (ADME) properties were analyzed using Qikprop in order to predict the pharmacologically relevant properties for both imatinib alone and PST-IM complex, as depicted in Table 5.
ADME prediction for both the drug “imatinib” and drug complexed with the monomer of polysaccharide.

Parameters such as QPlogBB, donor HB, QPlogS, QPlogP0/w, accept HB satisfied the recommended values for the drug. But in the case of drug polysaccharide complex, hydrogen bond acceptor got the value of 25.2. It is above the range recommended. Human oral absorption is medium for drug whereas low for imatinib–polysaccharide complex.
In vitro cytotoxicity
Since imatinib mesylate is reported to be a targeted drug with specific site of action on BCR/ABL oncoprotein, the PST-IM nanoparticles were also evaluated for their anticancer potential on various cancer cell lines. This potential was found to be highly significant (p < 0.001) in Ph-positive K562 cell line and K562 resistant cell line while there was no significant cytotoxicity in Ph-negative Jurkat and HL-60 cell line. Cytotoxicity of imatinib mesylate, PST001, and PST-IM nanoparticles was also evaluated in leukemic cell lines for 24, 48, and 72 h. BCR/ABL positive leukemic cell line K562 and resistant K562 were growth-arrested with IC50 values of 0.001 µg/mL and 1 µg/mL, respectively, after 48 h of incubation with the nanoparticles, whereas the imatinib mesylate achieved IC50 of 0.9 µg/mL in K562 and failed to inhibit 50% of cell growth in K562R even at a higher concentration and with a longer incubation period (Figure 10). The cytotoxic potential of the nanoparticles increased in a dose-dependent manner in both cell lines. PST001 achieved no cytotoxicity in leukemic cell lines even after longer incubation periods, whereas when imatinib was used in combination with PST001, the cytotoxic activity of imatinib was slightly enhanced (Supplementary Information Figure 1). PST-IM nanoparticles exhibited a dose-dependent increase in cytotoxicity in K562 cells but reached optimum cytotoxicity at 1 µg/mL in K562R cells at 24 h of incubation (Figure 10). To evaluate the cytotoxic potential of PST001 in BCR/ABL negative cell lines, an MTT assay was performed on Jurkat and HL-60 cell lines. PST-IM nanoparticles were found not to be cytotoxic to the cells across all concentrations indicating its specific target via fusion oncoprotein (Supplementary Information Figure 2). Various imatinib formulations were previously reported to have excellent anticancer effect;6–8 however, this study revealed that PST-IM nanoparticles are more potential than free drug. Also, it was reported first time about the use of imatinib-encapsulated biopolymer-coated nanoparticles that reverses chemo-resistance developed in leukemic cell lines.
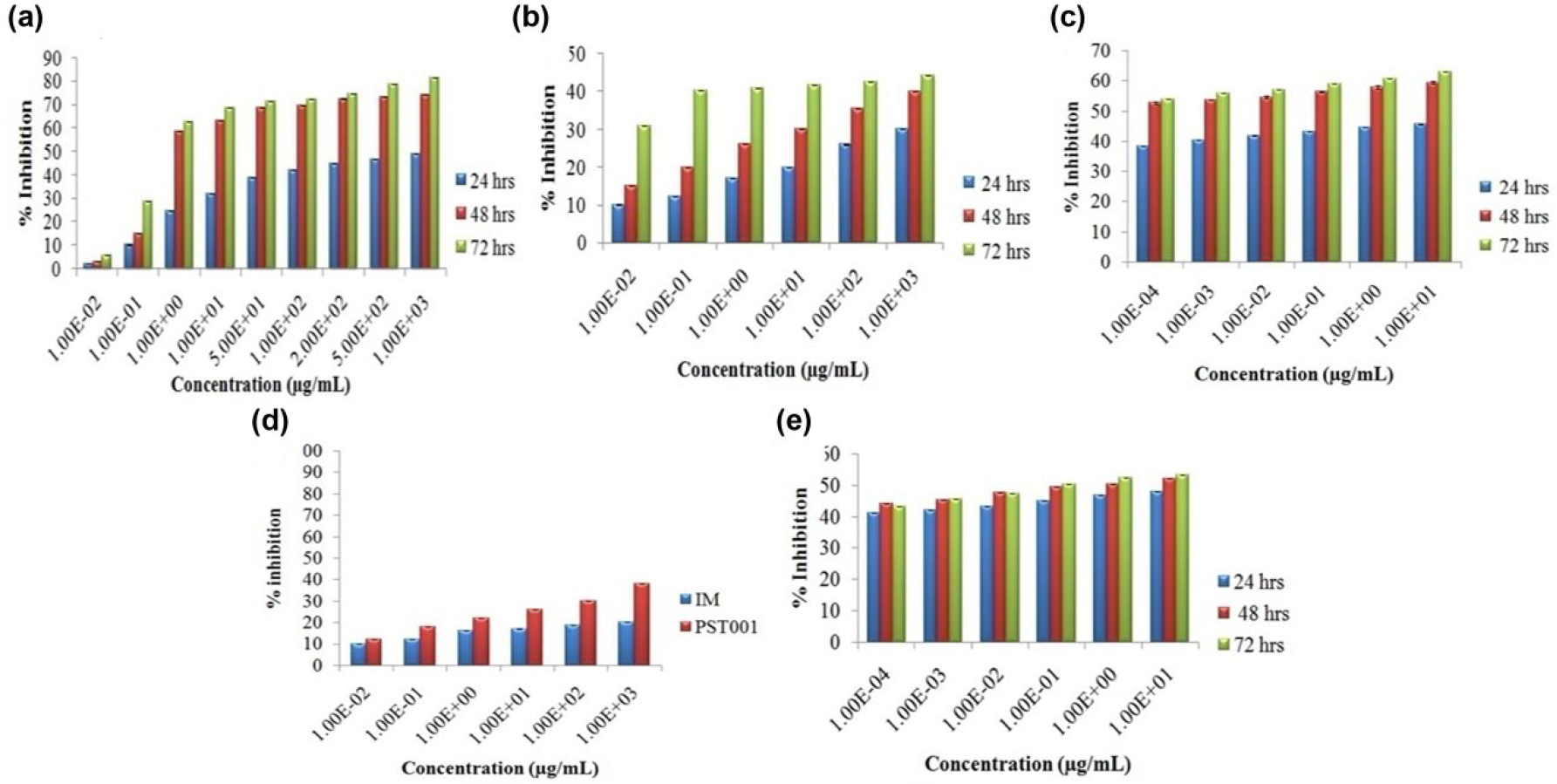
Assay for cytotoxicity in cancer cell lines treated with agents. Cytotoxicity profiling of K562 cells with (a) imatinib, (b) PST001, and (c) PST-IM nanoparticles cells at 24, 48, and 72 h. Cytotoxicity profiling of K562R cells with (d) imatinib and PST001 for 48 h and (e) PST-IM nanoparticles cells at 24, 48, and 72 h. Results are expressed as mean ± SD.
PST-IM nanoparticles exhibit excellent apoptosis in resistant Ph-positive cell lines compared to free drug
Because PST-IM nanoparticles exhibited significant cytotoxicity specifically against K562 and K562R cell lines, their mode of cell death induction was evaluated using various apoptotic assays. Membrane blebbing, which is a hallmark of apoptosis, refers to irregular bulges in the plasma membrane of a cell caused by localized decoupling of the cytoskeleton from the plasma membrane. Morphological evaluation of the cell lines treated with PST-IM nanoparticles at a concentration of 1 µg/mL for 48 h using phase contrast microscopy revealed a decrease in the number of cells exhibiting morphological features of apoptosis, such as distorted shape and membrane blebbing in comparison with the control group (Figure 11). Using acridine orange–ethidium bromide staining, cells treated with PST-IM nanoparticles showed a change in color from green to yellow/red with associated apoptotic features compared to the negative control (Figure 11). The Hoechst nuclear staining of negative control cells had intact round nucleus and normal morphology. When the cells were treated with free drug and PST-IM nanoparticles for 48 h, exhibited a bright blue color emission, concluding the nuclear fragmentation with increased chromatin condensation leading to induction of apoptosis. The enhanced activity exhibited by the nanoparticles compared with the parent drug might be due to the increased uptake of the particles via endocytosis because of their smaller size and increased surface to volume ratio.
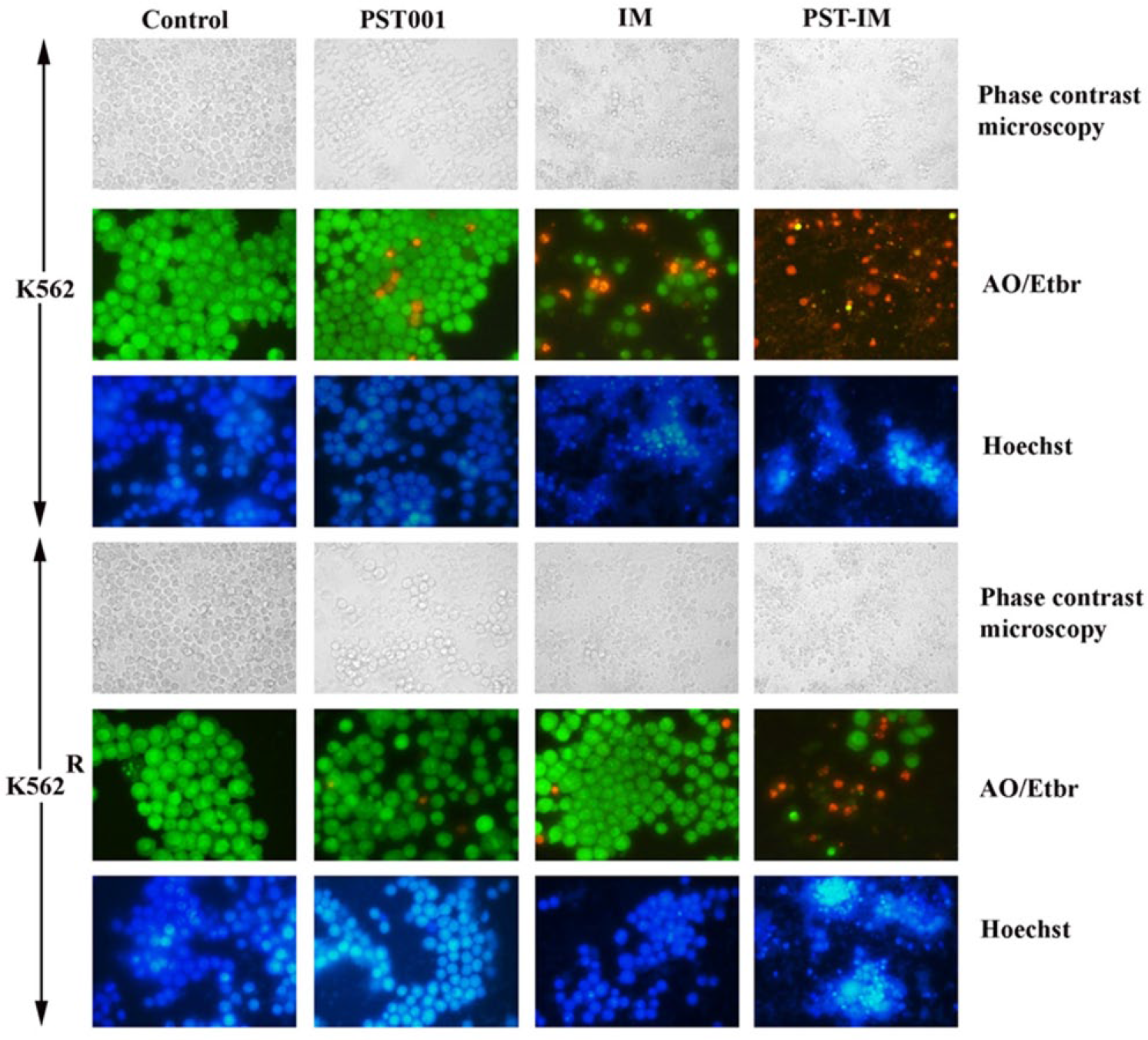
Apoptotic evaluation of K562 and K562R cell lines treated with PST001, imatinib, and PST-IM nanoparticles for 48 h. The morphological analysis was carried out using phase contrast microscopy, live–dead staining using acridine orange/ethidium bromide (AO/Etbr) stain, and nuclear condensation studies using Hoechst staining.
Gene expression analysis
The effect of PST-IM on the status of the drug transporter Pgp was studied in K562 and K562R cells at both transcriptional and translational level. PST-IM nanoparticles could not effectively reduce the expression of Pgp in K562R cells. Although Pgp levels are unaltered, the resistance in K562 cells was reversed by PST-IM as evident from cytotoxicity studies (Figure 12). This may be attributed to its small size which can easily evade the efflux pump. However, further studies are required to confirm the mechanism of action of PST-IM in resistance reversal.

Gene and protein expression analysis of Pgp in K562 and K562R cell lines.
In vivo toxicity
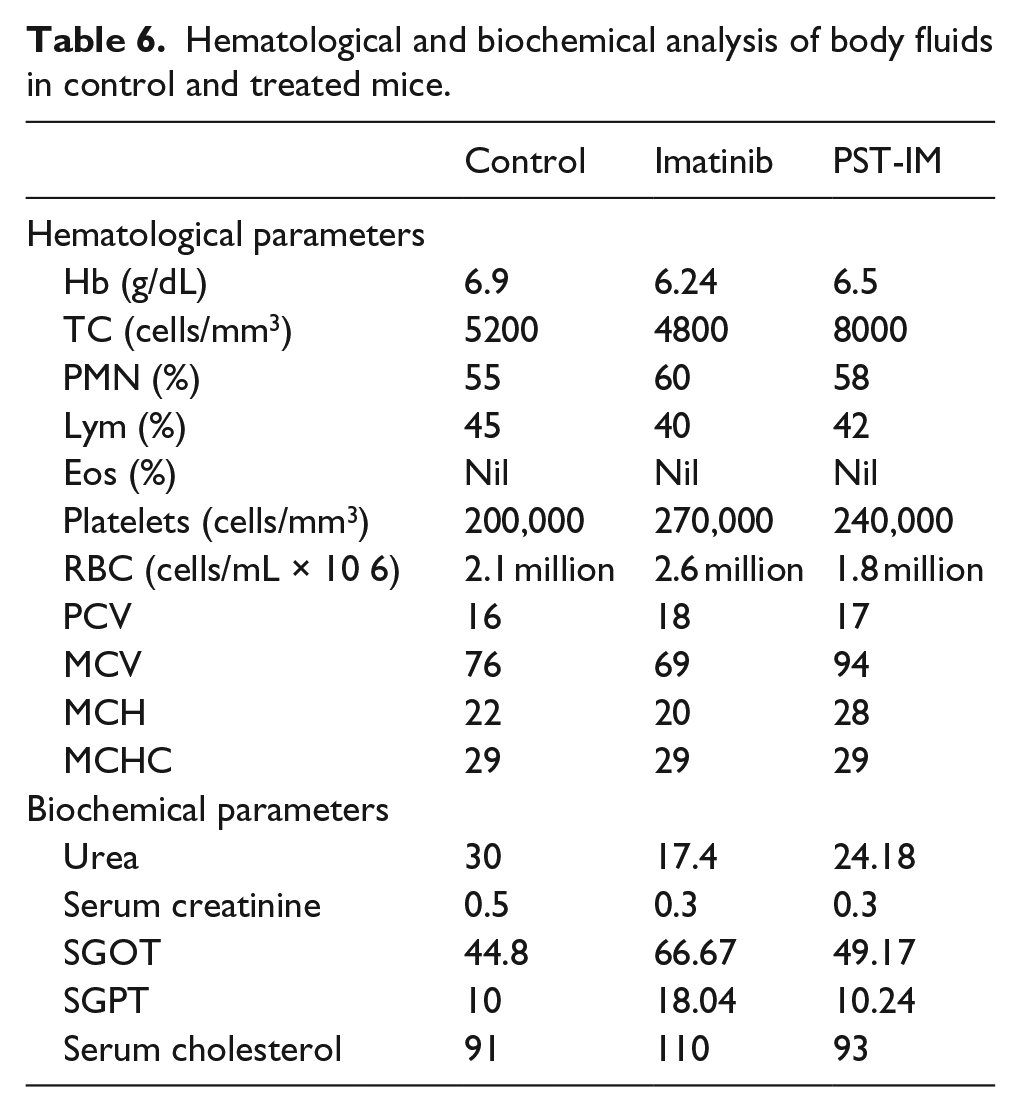
The results of hematological and biochemical analysis of blood and serum collected from mice administered with PST-IM nanoparticles were tabulated in Table 6. There was a dose-dependent decrease in the total white blood cells (WBC), red blood cells (RBC), and Hb counts in the imatinib alone group, while the 50 mg/kg PST-IM treated groups showed lower Hb and RBC, but increased WBC levels. The kidney function indicators, urea and creatinine, and liver function indicators, SGOT and SGPT, were significantly altered in the IM treated groups and in the PST-IM treated group. Hematoxylin and eosin staining were performed on the liver, kidney, lung, heart, and spleen of the sacrificed animals to evaluate any pathological changes associated with the administration of the compounds at the organ level.
Hematological and biochemical analysis of body fluids in control and treated mice.
Histopathological evaluation
Histopathological analysis of various organs from mice after administration of PST-IM nanoparticles revealed no significant toxicity-induced damage compared to those treated with free drug. This is in agreement with the fact that encapsulation of chemotherapeutic drugs including imatinib mesylate as nanoparticles will overcome drug-induced organ toxicity. 19 Administration of free drug in the dose of 50 mg/kg showed pathological changes like inter-fibrillar hemorrhage, disruption of cardiac muscle, and focal myolysis in heart tissue which minimized in those groups treated with PST-IM nanoparticles (Figure 13; Supplementary Information Tables 1–3). No significant pathological changes were observed in lungs and spleen in PST-IM nanoparticles–treated group; however, slight edema and bronchial cell hyperplasia were observed in lungs with those treated with drug (Supplementary Information Tables 4 and 5). Nephrotoxicity and hepatotoxicity observed in drug-treated animal group significantly minimized in nanoparticle-treated group. Thus, PST-IM nanoparticles reduce the toxicity induced by free drug in various organs and thus could be a safer delivery platform in leukemia patients with drug resistance.

Histopathological investigation of various organs from mice orally treated with imatinib mesylate free drug and PST-IM nanoparticles (50 mg/kg) for consecutive 28 days.
Discussion
Imatinib mesylate serves as a model drug with tyrosine kinase–related abnormalities and targeted therapy in general. 20 It is used to treat different types of leukemia, in particular, Philadelphia chromosome positive chronic myelogenous leukemia (CML), as well as gastrointestinal stromal tumors and other types of cancer. Cancer chemotherapy with imatinib is often associated with side effects including diarrhea, nausea, muscle pain, and fatigue. 20 In this current investigation, we have evaluated the potency of a nanoconjugate of imatinib with PST001 to improve the effectiveness of therapy by overcoming multidrug resistance.
Despite the fact that various studies have reported, efficacy of various formulations of imatinib, this study is first of its kind to use imatinib encapsulated within a biopolymer. The nanoparticles were prepared using galactoxyloglucanas, a polymer, and by using DMSO–water gelation method along with copolymer PVA. Previous studies proved that the co-encapsulation of doxorubicin with PST001 exhibited minimal cardiotoxicity and significant tumor specific cytotoxicity. 18 The prepared nanoparticles exhibited an effective encapsulation with appreciable drug payload. The swelling behavior of nanoparticles in physiological pH favors the release of drug in controlled behavior in body fluids that was indicated by drug release of 63% total encapsulated imatinib mesylate during the first 24 h of incubation at pH 7.4.
We next examined the docking score for finding the protein–ligand interactions in molecular docking of free drug and PST-IM conjugate. Here, we used induced fit docking method (flexible docking) using glide tool available in Schrodinger. Imatinib was docked with three different proteins such topoisomerase, EGFR tyrosine kinase domain, and ABL1 kinase domain. The binding affinity of drug–polysaccharide complex shows better result rather than the drug alone with all these proteins. PST-IM conjugate shows highest dock score in the docking with topoisomerase. In the case of docking of drug alone with different proteins, the better interaction results in docking with EGFR tyrosine kinase domain proteins. We also examined the docking study between the model of polysaccharide and these proteins. These results also show better interaction with topoisomerase.
PST-IM nanoparticles exhibits significant cytotoxicity to K562 cells compared to imatinib mesylate after 48 h of incubation. The cytotoxicity was achieved in K562R after 48 h of incubation of nanoparticles while cell viability was not significantly decreased on treatment with free drug. After 72 h of treatment, PST-IM showed 60% growth inhibition in resistant K562 cells whereas imatinib mesylate did not reach even the IC50. This shows the advantage of developing nanoconjugates of imatinib with biopolymers like polysaccharides for prolonged bioavailability of imatinib for better efficacy. The overexpression of Pgp efflux pumps considered to a major factor for the resistance of cells. The size of imatinib nanoparticles may be too large to be effluxed by Pgp. Therefore, compared with imatinib mesylate, nanoparticles show enhanced inhibition of drug-resistant cancer cells. The nanoparticles exhibited no significant cytotoxicity for BCR/ABL negative leukemia cell lines (HL-60 and Jurkat) that demonstrates the targeted inhibition of BCR/ABL fusion protein. The molecular mechanisms involved in the cytotoxicity of PST-IM in resistant cells has to be investigated further, as our studies revealed that PST-IM could not reduce the expression of Pgp either at the transcriptional level or at the translational level. One of the possibilities could be that the nanoconjugate directly inhibits the function of the Pgp transporter without altering its expression. Numerous studies support the inhibition of Pgp function without altering its expression. Studies by Mi and Lou 21 on the ability of ZD6474 in reversing Pgp-mediated drug resistance showed enhanced uptake and/or decreased efflux in resistant cells without affecting the expression of Pgp. The in vivo toxicity studies revealed that the nanoconjugate has no evident toxic effects which hold up the efficacy of PST-IM over the parent compounds. There are mainly three generations of Pgp modulators among which the first-generation modulators inhibit the activity of ABCB1/Pgp, but its low binding affinity and high toxicity at doses required for multidrug resistance (MDR) reversal limited its clinical benefits. The second-generation ABC modulator, valspodar reduces the effective doses of other concomitantly used anticancer agents due to complex pharmacokinetic interactions, so its clinical application is limited.22,23 Elacridar, a dual ABCB1/Pgp and ABCG2/BCRP inhibitor and third-generation ABC modulator, exhibits significant MDR reversal effect with only minimal side effects. 24 Recently, much effort has been undertaken to identify or synthesize selective modulators of ABC transporters with limited nonspecific toxicity. Thus, PST-IM can be considered an effective nanoparticle-based drug delivery system that could reverse drug resistance with minimal side effects.
Conclusion
The blind use of imatinib is limited due to the side effects and development of drug resistance. This study focused on the encapsulation of polysaccharide on imatinib to improve the effectiveness and biocompatibility. Tamarind seed polysaccharide acts as a carrier to deliver the drug to its target site to minimize side effects. In vitro studies showed that the nanoconjugate of imatinib mesylate with PST001 could effectively enhance the cytotoxic potential of imatinib and could reverse the multidrug resistance in K562R cell line. The drug could not, however, alter the expression of Pgp, a major factor that contributes to MDR. PST-IM is a better cytotoxic agent than both of its parent counterparts as evidenced by in vitro and in silico studies and is effective even in resistant cells. Lack of any marked in vivo toxicity makes it even more appealing for further pre-clinical studies.
Footnotes
Acknowledgements
B.S.U., G.U.P., and R.S. thank University Grants Commission (UGC), Govt. of India for research fellowship (UGC-JRF). M.M.J. thank Kerala Biotechnology Commission (KBC-KSCSTE), Govt. of Kerala for Post-Doctoral Research Fellowship. A.R.J. and B.S.U. equally contributed.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors thank Kerala State Council for Science, Technology and Environment for financial support for the work (No. 013/SRSHS/2013/CSTE dated 11/04/2014). A.R.J. thanks Council of Scientific and Industrial Research (CSIR) for the Junior Research Fellowship (No: 09/553(0021)/2010-EMR-1 dated 22/12/2010).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.