
Abstract
Pancreatic cancer is one of the most aggressive and difficult to treat cancers. Experimental and clinical evidence suggests that high basal state autophagy in pancreatic tumors could induce resistance to chemotherapy. Recently, we have demonstrated that penfluridol suppresses pancreatic tumor growth by autophagy-mediated apoptosis both in vitro and in vivo; however, the mechanism of autophagy induction by penfluridol was not clear. Several studies have established that endoplasmic reticulum stress could lead to autophagy and inhibit tumor progression. In this study, we demonstrated that penfluridol induced endoplasmic reticulum stress in BxPC-3, AsPC-1, and Panc-1 pancreatic cancer cell lines as indicated by upregulation of endoplasmic reticulum stress markers such as binding protein (BIP), C/EBP homologous protein (CHOP) and inositol requiring 1α (IRE1α) after treatment with penfluridol in a concentration-dependent manner. Inhibiting endoplasmic reticulum stress by pretreatment with pharmacological inhibitors such as sodium phenylbutyrate and mithramycin or by silencing CHOP using CHOP small interfering RNA, blocked penfluridol-induced autophagy. These results clearly indicate that penfluridol-induced endoplasmic reticulum stress lead to autophagy in our model. Western blot analysis of subcutaneously implanted AsPC-1 and BxPC-3 tumors as well as orthotopically implanted Panc-1 tumors demonstrated upregulation of BIP, CHOP, and IRE1α expression in the tumor lysates from penfluridol-treated mice as compared to tumors from control mice. Altogether, our study establishes that penfluridol-induced endoplasmic reticulum stress leads to autophagy resulting in reduced pancreatic tumor growth. Our study opens a new therapeutic target for advanced chemotherapies against pancreatic cancer.
Keywords
Introduction
Pancreatic cancer is the fourth leading cause of death due to cancer and is also associated with lowest survival rate among any other solid tumor. 1 Despite in-depth understanding of its pathology and biology, current therapies do not increase the overall survival of pancreatic cancer patients. 2 Profound chemoresistance of pancreatic cancer has been associated with limited benefit to the patients receiving standard chemotherapy. 3 Endoplasmic reticulum (ER) is an organelle, which is mainly involved in the synthesis of protein, modification and folding. A number of physiological conditions such as oxidative stress and glucose deprivation disrupt folding of proteins in ER. 4 Accumulation of misfolded proteins in ER lumen leads to ER stress. 5 ER stress represents a cellular process, where low to moderate stress supports cell growth and chemoresistance, whereas high ER stress turns on cell death program. 6 It has been shown that potential therapeutic response can be achieved by inducing ER stress. 7 Pancreatic cancer cells have prominent ER, hence may represent attractive therapeutic target. 7
Antipsychotic drugs such as olanzapine and risperidone are known to induce ER stress. 8 Furthermore, olanzapine was also observed to induce marked apoptosis in pancreatic β cells. 8 It has been demonstrated that Withaferin A, a biologically active withanolide extracted from Withania somnifera induces apoptosis in pancreatic cancer through ER stress-mediated autophagy. 9 In another study, penfluridol was shown to inhibit pancreatic cancer by activation of protein phosphatase 2A. 10 Recently, we have demonstrated that penfluridol, an antipsychotic drug, suppresses pancreatic tumor growth by autophagy-mediated apoptosis. 11 However, the mechanism leading to penfluridol-induced autophagy in pancreatic cancer remains unknown.
Interaction between autophagy and ER stress in cancer remains unexplained.12,13 Nonetheless, role of ER stress and autophagy has been established in cancer progression and chemoresistance6,14,15 In this study, we observed that penfluridol treatment induced ER stress both in vitro and in vivo in pancreatic cancer. Our results also demonstrated that penfluridol-induced ER stress leads to autophagy resulting in pancreatic tumor growth suppression. To the best of our knowledge, this is the first report establishing relationship between penfluridol-induced ER stress and autophagy in pancreatic cancer.
Methods
Ethics statement
Experiments in mice were conducted in accordance with the ethical standards and according to approved protocol by Institutional Animal Care and Use Committee (IACUC), Texas Tech University Health Sciences Center, Amarillo, TX, USA.
Cell culture
AsPC-1 and BxPC-3 human pancreatic cancer cell lines were obtained from American Type Culture Collection (ATCC), Manassas, VA, USA. Panc-1 cells were provided as a kind gift from Dr Thomas L. Brown (Wright State University, Dayton, OH, USA). All the cell lines used in the study were authenticated using short tandem repeat (STR) method at institutional core laboratory. Cells were cultured and maintained as we have described previously. 16 All the pancreatic cancer cell lines used in this study were within 20 passages after receipt or resuscitation.
Western blotting
Panc-1, AsPC-1, and BxPC-3 cells were treated with 2.5, 5, 7.5, and 10 µM penfluridol (concentration-dependent manner) for 24 h. After treatment, cells were collected and washed with phosphate-buffered solution (PBS) twice. Cell lysis was performed and protein content was determined using Bradford reagent and as described by us previously. 16 After protein estimation, 20–60 µg of protein was subjected to sodium dodecyl sulfate (SDS) gel electrophoresis and resolved proteins were transferred to polyvinylidene fluoride (PVDF) membrane followed by immunoblotting as described by us previously. 16 The membranes were probed for primary antibodies against binding protein (BIP), C/EBP homologous protein (CHOP), inositol requiring 1α (IRE1α), LC3B, p62, as well as actin. All the primary antibodies were procured from Cell Signaling Technology (Danvers, MA) except actin which was purchased from Sigma Aldrich (St. Louis, MO). The membranes were developed as described by us previously. 17
Inhibitors pretreatment
A volume of 0.3 × 106 AsPC-1 cells were plated per well in a six-well plate. After overnight incubation, cells were pretreated for 3 h with 5 mM sodium phenylbutyrate and 100 nM mithramycin. Sodium phenylbutyrate and mithramycin were purchased from Sigma Aldrich, St. Louis, MO, USA. After pretreatment, 5 µM penfluridol was added for 24 h. Cells were collected and processed for western blotting as explained above.
Silencing of CHOP
CHOP small interfering RNA (siRNA; Cell Signaling Technology, Danvers, MA, USA) was used to silence CHOP in AsPC-1 cells using siPORT (Ambion, Inc., Austin, TX, USA) transfection reagent as per manufacturer’s protocol. Cells were transfected with 100 nM CHOP siRNA or scrambled siRNA. After 24 h post transfection, cells were treated for additional 24 h with 5 µM penfluridol. The cells were collected after treatment with penfluridol and processed for western blot analysis as described by us before. 11
Subcutaneous implantation of pancreatic tumor
Athymic nude mice 4–6 weeks old were purchased from Harlan Laboratory (Livermore, CA, USA). An amount of 1 × 106 AsPC-1 and BxPC-3 cells in 1:1 mixture of PBS and Matrigel were implanted in right and left flank of mice, respectively. Once tumor volume was around 70 mm3, mice were randomly divided with five mice in each group. Group I served as control and received the vehicle only. Group II received 10 mg/kg penfluridol by oral gavage every day. Tumor volume was measured twice a week till day 27 using vernier caliper as described by us before.11,17 Mice were humanely sacrificed and tumors were removed. The results of this experiment have been published. 11 A part of tumors from control- and penfluridol-treated mice from this experiment was used to perform western blot analysis in this study.
Orthotopic tumor model
Female athymic nude mice 4–6 weeks old from Harlan Laboratory were used for orthotopic implantation of tumors. Isoflurane was used to anesthetize the mice, and a small incision was made to implant stably luciferase-expressing Panc-1 (Panc-1-luc) cells orthotopically in the pancreas. PBS suspension containing 1 × 106 exponentially growing Panc-1-luc cells in 20 µL were injected into the subcapsular region on the pancreas using a 30-gauge sterile needle. The peritoneum and skin incisions were closed sequentially with absorbable suture. Pain killer was given to the mice at every 8 h for initial 2 days. On the day of injection, mice were imaged using IVIS (Caliper Biosciences, MA) to have basal luminescence value after injecting luciferin (3 mg/mouse, intraperitoneal (ip)). On day 9 after the surgical implantation of Panc-1-luc cells, mice were divided randomly into two groups with six mice in each group. A volume of 10 mg/kg penfluridol by oral gavage was provided to the treatment group, whereas control group received vehicle only. To measure tumor growth, tumor luminescence in pancreas was measured thrice a week. At the end of the experiment, mice were humanely sacrificed; pancreases with tumors were excised and snap frozen. Tumor growth curve in control and penfluridol-treated group obtained from this experiment has been published. 11 A part of snap-frozen orthotopic pancreatic tumor samples from control and penfluridol-treated group from this experiment was used to perform western blot analysis for ER stress markers in this study.
H&E staining
Formalin-fixed tumor tissues were dehydrated and paraffin embedded. Microtome (Leica Microsystems Inc., Buffalo Grove, IL, USA) was used to section tissues into 5-µm-thick sections. The sections were deparaffinized and rehydrated by performing three washes with xylene for 10 min each and two washes with 100% ethanol of 5 min each followed by two washes with 95% ethanol for 6 min and one wash each of 70% and 50% ethanol for 3 min. Sections were then given two 5-min washes in distilled water (dH2O) followed by staining with Mayer hematoxylin solution for 10 min. After washing, sections were counterstained in 0.25% eosin Y solution followed by dehydration and mounting.
Results
Penfluridol induces ER stress in pancreatic cancer
It has been established that autophagy can be activated by ER stress, eventually leading to apoptosis. 13 We have recently shown that penfluridol induces autophagy-mediated apoptosis in pancreatic cancer. 11 Hence, we wanted to investigate whether penfluridol induces ER stress in pancreatic cancer cells. Our results demonstrated that penfluridol treatment increased the expression of ER stress response molecules such as BIP, CHOP, and IRE1α in three different pancreatic cancer cell lines Panc-1, AsPC-1, and BxPC-3 after 24 h of treatment in a concentration-dependent manner indicating ER stress (Figure 1(a)–(c)). X-box binding protein 1 (XBP1) is another important ER stress response factor. However, in our study, we did not observe any significant change in XBP1 with penfluridol treatment in AsPC-1 (data not shown).

Induction of endoplasmic reticulum stress with penfluridol treatment. Panc-1, AsPC-1, and BxPC-3 cells were treated with different concentrations of penfluridol for 24 h. Representative blots showing concentration-dependent effect of penfluridol on BIP, CHOP, and IRE1α expression. Binding protein (BIP), C/EBP homologous protein (CHOP) and inositol requiring 1α (IRE1α) are ER stress markers. Figure includes representative blots of at least three independent experiments. Actin was used as loading control.
Blocking ER stress inhibits penfluridol-induced autophagy
To establish that ER stress induced by penfluridol leads to autophagy, we pretreated pancreatic cancer cells with sodium phenylbutyrate or mithramycin, which are well-known inhibitors of ER stress, followed by treatment of cells with 5 µM penfluridol. Our results showed that blocking ER stress suppressed induction of autophagy, suggesting that penfluridol-induced ER stress leads to autophagy in pancreatic cancer (Figure 2(a) and (b)).
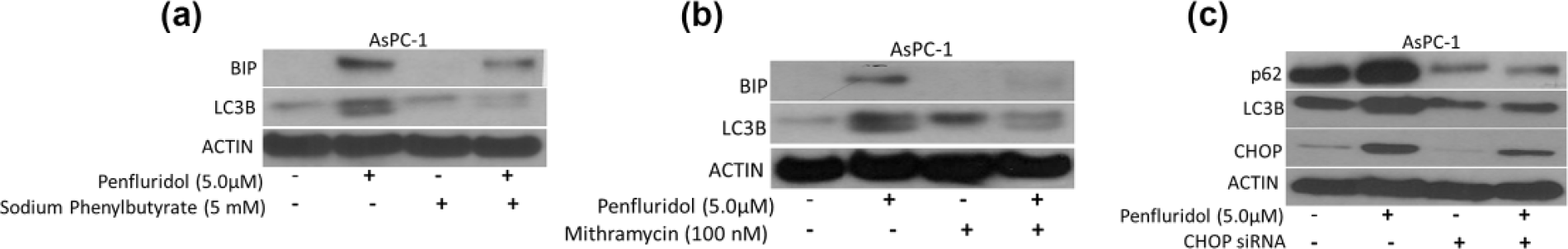
Penfluridol-induced endoplasmic reticulum stress leads to autophagy in pancreatic cancer. Approximately 0.3 × 106 AsPC-1 cells were plated in six-well plates. Cells were pretreated with (a) 5 mM sodium phenylbutyrate and (b) 100 nM mithramycin and (c) transfected with CHOP siRNA. Cells were treated with 5 µM penfluridol for 24 h. Levels of BIP, CHOP, LC3B, and p62 were evaluated by western blotting. Actin was used as loading control. Figure shown includes representative blots of at least three independent experiments.
Silencing of CHOP attenuated induction of autophagy by penfluridol treatment
In another experiment, CHOP was silenced using CHOP siRNA before treatment with penfluridol. Similar to our previous observations, we observed that silencing of CHOP resulted in inhibition of penfluridol-induced autophagy (Figure 2(c)). These results established the link between ER stress and autophagy in our model.
Penfluridol induced ER stress in subcutaneously implanted pancreatic tumors
In order to confirm the in vitro observations of penfluridol-induced ER stress, AsPC-1 and BxPC-3 tumors from control and penfluridol-treated mice from the previous study were analyzed for ER stress by western blotting. Our results showed that ER stress markers such as BIP, CHOP, as well as IRE1α were upregulated in the tumors from penfluridol-treated mice as compared to control mice indicating ER stress in vivo by penfluridol (Figure 3(a) and (b)).

Endoplasmic reticulum stress induced by penfluridol treatment in subcutaneously as well as orthotopically implanted pancreatic tumors. (a) AsPC-1 and (b) BxPC-3 subcutaneously implanted pancreatic tumors were removed after terminating the experiments. Tumors were homogenized, lysed, and analyzed for BIP, CHOP, and IRE1α. Binding protein (BIP), C/EBP homologous protein (CHOP) and inositol requiring 1α (IRE1α) are ER stress markers. (c) After terminating the experiment, pancreas with Panc-1 tumors were removed, lysed, and analyzed for CHOP and IRE1α by western blotting. Actin was used as loading control. Each lane of blot represents tumor from separate mouse.
Induction of ER stress by penfluridol treatment in orthotopically implanted pancreatic tumors
Induction of ER stress by penfluridol treatment was further confirmed in orthotopic pancreatic tumor model. The pancreatic tumors from a previously conducted orthotopic tumor study were taken for analysis. 11 Our results demonstrated that penfluridol treatment induced ER stress in orthotopically implanted Panc-1 tumors as indicated by increase in ER stress markers like CHOP and IRE1α (Figure 3(c)). H&E staining of AsPC-1 tumor tissues from control and penfluridol treatment revealed that cells in the treated tissue were less densely packed as compared to control tissue indicating that penfluridol is effective in suppressing tumor growth (Figure 4). Taken together, results from two different in vivo tumor studies clearly showed induction of ER stress in agreement with our in vitro observations.
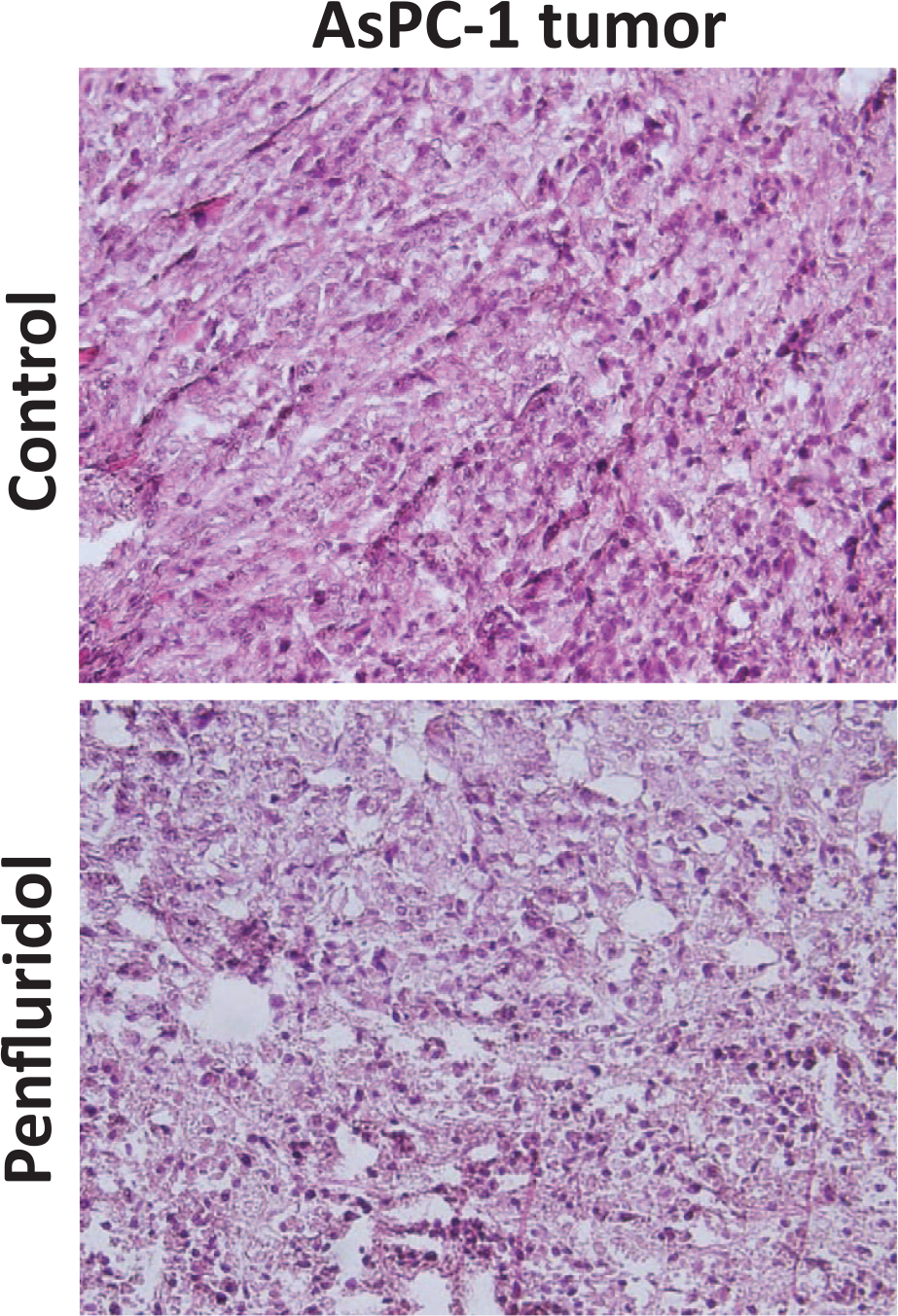
Change in tumor density associated with penfluridol treatment. Tumors were sectioned and H&E staining was performed as described in “Methods” section.
Discussion
Pancreatic cancer is one of the most lethal cancers due to resistance to standard therapies. 18 The 5-year survival rate of pancreatic cancer patients is less than 5%, making it a highly death-dealing malignancy. 19 Majority of pancreatic cancer patients are diagnosed at late stage, which is non-operable clinical stage. However, in patients with early diagnosis and successful surgical resection, tumors reoccur as locally advanced or metastatic cancer within 2–5 years. 19 Innovation in systemic therapy to treat pancreatic cancer patients is clearly necessary to overcome chemoresistance of pancreatic tumors and for robust and sustained survival outcome.
ER is the central intracellular organelle, which is involved in secretory function as well as folding, translocation, and post-translocation modifications of the secreted proteins. Perturbation in ER function is accompanied by accumulation of unfolded protein, a process termed as ER stress. Several experimental evidences have suggested that ER stress induces cellular dysfunction and cell death, making ER stress induction an attractive strategy for therapeutics. 20 Interestingly, antipsychotic drugs such as clozapine, haloperidol, olanzapine, and risperidone are known to induce ER stress.8,21 ER stress has been linked to induce autophagy-mediated cell death in cancer. Several antipsychotic drugs such as fluspirilene, trifluoperazine, and pimozide are known to induce autophagy. 22 Penfluridol has been shown to activate protein phosphatase 2A and inhibit pancreatic cancer. 10 Recently, we have demonstrated that penfluridol, an antipsychotic drug, induced autophagy-mediated apoptosis and pancreatic tumor growth suppression. 11 However, the mechanism behind penfluridol-induced autophagy in pancreatic cancer is unknown. In this study, we investigated the cellular machinery responsible for induction of autophagy upon treatment with penfluridol. Interestingly, several epidemiological studies have established that patients taking neuroleptics are less prone to cancer. 23 According to an in vitro study conducted at University of New South Wales, pimozide inhibits lung, breast, and brain cancer. In our previous study, we have established the anti-cancer effects of penfluridol against breast cancer and its metastasis to brain through inhibition of integrin signaling, whereas Wu et al. 24 observed dysregulation of cholesterol homeostasis by penfluridol in breast cancer. 17
ER plays critical role in synthesis and secretion of transmembrane and secretary proteins. The adaptive response to ER stress is unfolded protein response (UPR). 25 The UPR is initiated by ER transmembrane proteins such as IRE1α. Under normal or unstressed condition, ER chaperone and immunoglobulin BIP bind to the luminal domain of IRE1α and keep them inactive. However, during ER stress, these proteins dissociate from IRE1α making it active. 25 CHOP was reported to be involved in ER stress–mediated apoptosis through IRE1-induced transcriptional stimulation.4,26 Our results demonstrated that penfluridol-mediated ER stress was associated with increase in BIP, CHOP, and IRE1α in vitro and in vivo. Inhibiting ER stress by pretreatment with sodium phenylbutyrate or mithramycin or by silencing CHOP using siRNA for CHOP resulted in blocking penfluridol-induced autophagy in pancreatic cancer, indicating direct regulation of ER stress–mediated autophagy in our model. Supporting our findings, several other anti-cancer agents have been shown to induce ER stress, resulting in direct or indirect effects on tumor growth. 27 Induction of ER stress reduces adaptation of cells to hypoxia, inflammation, and angiogenesis resulting in tumors getting sensitive to therapies. 28
Autophagy is a lysosome-dependent intracellular process, which degrades unwanted intracellular organelle and misfolded proteins. 22 It has also been observed that reduction of autophagy leads to accumulation of misfolded proteins. 22 The role of autophagy in cancer progression is highly controversial. Interestingly, induction of autophagy has been established to inhibit tumorigenesis, whereas autophagy induction is also known to support tumor growth. 29 We have recently established that penfluridol suppresses pancreatic tumor growth by autophagy-mediated apoptosis. 11 In this study, we demonstrated that autophagy by penfluridol was mediated through ER stress in pancreatic cancer.
This is the first report to establish that penfluridol-induced ER stress leads to autophagy resulting in pancreatic tumor growth suppression. ER stress and autophagy could be the potential targets for new and advanced chemotherapies. Our study established the link between ER stress and autophagy, laying the foundation for effective treatment options for pancreatic cancer.
Footnotes
Declaration of conflicting interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by R01 grant CA129038 (to Sanjay K Srivastava) awarded by the National Cancer Institute, NIH.