
Abstract
We provide herein several lines of evidence to substantiate that folic acid (or folate) is a micronutrient capable of functioning as a novel redox regulator on hepatocellular carcinoma. First, we uncovered that folate deficiency could profoundly downregulate two prominent anti-apoptotic effectors including survivin and glucose-regulated protein-78. Silencing of either survivin or glucose-regulated protein-78 via small interfering RNA interfering technique established that both effectors could serve as reactive oxygen species sinker proteins. Second, folate deficiency–triggered oxidative–nitrosative stress could strongly induce endoplasmic reticulum stress that in turn could provoke cellular glutathione depletion through the modulation of the following two crucial events: (1) folate deficiency could strongly inhibit Bcl-2 expression leading to severe suppression of the mitochondrial glutathione pool and (2) folate deficiency could also profoundly inhibit two key enzymes that governing cellular glutathione redox regulation including γ-glutamylcysteinyl synthetase heavy chain, a catalytic enzyme for glutathione biosynthesis, and mitochondrial isocitrate dehydrogenase 2, an enzyme responsible for providing nicotinamide adenine dinucleotide phosphate necessary for regenerating oxidized glutathione disulfide back to glutathione via mitochondrial glutathione reductase. Collectively, we add to the literature new data to strengthen the notion that folate is an essential micronutrient that confers a novel role to combat reactive oxygen species insults and thus serves as a redox regulator via upregulating reactive oxygen species sinker proteins and averting mitochondrial glutathione depletion through proper maintenance of redox homeostasis via positively regulating glutathione biosynthesis, glutathione transporting system, and mitochondrial glutathione recycling process.
Keywords
Introduction
Folic acid (folate; vitamin B9) plays a crucial role in DNA synthesis via the transfer of single carbon units in the biosynthesis of thymidylate and purine. 1 Even more importantly, folate, being a methyl group donor, also fulfills a cardinal role in the remethylation of homocysteine (Hcy) to methionine catalyzed by a B12-dependent methionine synthetase. 1 In an episode of folate deficiency (FAD), remethylation of Hcy can be blocked. Meanwhile, the failure of the transsulfuration reaction involving the conjugation of Hcy and serine to form cystathionine by a B6-dependent cystathionine β-synthase (CβS) will result in the accumulation of Hcy leading to the plethoric production of various reactive oxygen species (ROS) such as hydrogen peroxide (H2O2).2,3 FAD in tandem with dualistic deficiencies in vitamins B6 and B12 has been implicated as the etiological factor in the pathogenesis of various chronic degenerative diseases including cancer, diabetes, cardiovascular disorders, and Alzheimer’s disease, and free-radical-instigated oxidative/nitrosative stress is considered to be the root cause for the occurrence of these chronic degenerative diseases.4–6 This notion has been reasoned by Joshi et al., 7 and they present the first evidence to substantiate that folate may indeed possess free-radical-scavenging properties including CCl3O2• (a model peroxyl radical), N3•, SO4•—, and Br2•— (one electron oxidants) and physiological relevant OH• (hydroxyl) and O•— radical under ambient condition at almost a diffusion-controlled rate.Furthermore, these authors also demonstrated that folate could serve as a Fenton-modulator which might contribute indirectly for suppressing hydroxyl radical formation. 8 They therefore concluded that folate was an antioxidant based on its capability to effectively scavenge various types of free radicals. However, the question as to whether or not this micronutrient could also act as antioxidant through the indirect modulation of alternative antioxidative pathways has not been delineated.
Recently, we have utilized FAD-induced oxidative–nitrosative stress (ONS) system as a research platform to study the effects of ONS on the metastatic proclivity and drug resistance of hepatoma cells. Using this research platform, we were able to identify two phenotypes, including redox adaptation-prone (RAP) hepatocellular carcinoma (HCC) subclone variant such as Mahlavu cells and redox adaptation-null (RAN) HCC subclone variant such as Hep G2 cells, the former being able to initiate prosurvival pathways including overexpression of glucose-regulated protein-78 (GRP-78) and survivin leading to the evasion of apoptosis and conferring drug resistance attribute. Conversely, RAN phenotype (Hep G2 cells) was found to be devoid of this unique attribute and could lead to the apoptotic lethality. 9 Additionally, during the course of this investigation, we also uncovered that folate could play pivotal role in the positive regulation of some anti-apoptotic proteins in HCC RAP cells. By utilizing small interfering RNA (siRNA) knockdown technique, we proved for the first time that these anti-apoptotic proteins could act as ROS sinker. Most importantly, we also demonstrated that folate was a major effector in modulating mitochondrial redox homeostasis via regulating cytosolic glutathione (GSH) biosynthesis, influencing GSH transport system and modulating intracellular redox couples including GSH/glutathione disulfide (GSSG) and nicotinamide adenine dinucleotide phosphate (NADPH)/NADP+ which is essential for the mitochondrial GSH recycling process mediated by mitochondrial GSH reductase. Based on these data, we conclude that folate itself is a superb redox regulator.
Materials and methods
Materials
Unless otherwise specified, all chemical compounds including folate, nucleosides, amino acids, and nucleotides were purchased from Sigma-Aldrich (St Louis, MO, USA). The folate-deficient (FAD) medium without deoxyriboside, ribosides, deoxyribotides, ribotides, glycine, serine, and folate was formulated by Invitrogen and JRH (Lenexa, KS, USA). Penicillin, trypsin, fungizone, streptomycin, and Trypan blue were obtained from Gibco Laboratories (Grand Island, NY, USA). Fetal bovine serum (FBS) for cell culture was purchased from Biological Industries (Kibbutz, Israel). Antibodies used including COX-2, γ-glutamylcysteine synthetase heavy chain (γ-GCSh), Bcl-2, and GRP-78 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Survivin, β-catenin (Epitomics, Burlingame, CA, USA), pro-caspase 3 (Cell Signaling Technology, Danvers, MA, USA), and β-actin (Sigma-Aldrich) were obtained.
Cell line and cell culture
Human HCC cells including Hep G2 and Mahlavu subclone variants were obtained from the National Development Center for Biotechnology (Taipei, Taiwan) and used as the cell models. The minimal essential medium/alpha modified (αMEM) was a complete medium containing folate (2 µmol/L), hypoxanthine (36 µmol/L), thymidine (36 µmol/L), glycine (600 µmol/L), and serine (250 µmol/L), which was used to culture both cell types in the presence of 10% heat-inactivated FBS in a humidified atmosphere with 5% CO2 at 37°C. To eliminate contamination, penicillin (20,000 units/L), fungizone (2.5 mg/L), and streptomycin (20 mg/L) were also added to the culture medium. To prepare FAD media, two extra procedural steps are required. First, folate, thymidine, hypoxanthine, and glycine were omitted from the complete media to stress substrate availability in one-carbon metabolism. Second, FBS was dialyzed at 4°C for 16 h against 6 × 10 volumes of sterile phosphate-buffered saline (PBS; referred to as dialyzed FBS (dFBS)) in order to eliminate exogenous folate sources. Thus, HCC cells cultured in FAD medium (in the absence of folate and thymidine, glycine, hypoxanthine, and serine) and 10% dFBS are designated as FAD cells. HCC cells grown in control medium and 10% FBS are referred to as control cells (FAC).
Measurement of intracellular ROS and nitric oxide
The production of intracellular ROS/nitric oxide (NO) was detected either by flowcytometry or by confocal imaging microscopy using DCF-DA or DAF-FM probes. HCC cells were grown under FAC or FAD conditions for various time periods until 80% confluency was reached. The culture medium was then replaced with new medium and treated with 10 µM DCF-DA or DAF-FM for 30 min in the dark, washed once with PBS, detached by trypsinization, collected by centrifugation, and resuspended in PBS. The intracellular ROS or NO, as exhibited by the fluorescences of DCF or DAF trapped inside the cells, were measured with a Becton-Dickinson FACSCalibur flow cytometer. In addition, for intracellular ROS measured by confocal imaging microscopy, the fluorescent probes were loaded at 37°C in the dark for 30 min. After loading, the cells were rinsed three times with 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-buffered saline. Confocal fluorescence images were obtained using a Leica SP2 MP (Leica-Microsystems, Mannheim, Germany) fiber coupling system equipped with a Ti:Sa-Laser system (model:Millenia/Tsunem; Spectra-Physics; Mountain View, CA, USA) providing pulse repetition rate at 82 MHz, laser pulse width of 1.2 ps, spectral bandwidth of 1 nm, and object pulse width of 1.3 ps. Wavelength at 800 nm with average laser power of 600 mW was selected for illumination. During fluorescence imaging, the illumination light was reduced to the minimal level by selecting suitable neutral density filters to prevent the photosensitizing effect from the interaction of light with the fluorescence probes. All images were processed and analyzed using Leica QWin software (Leica Imaging System Ltd., Clifton Road, Cambridge, England). Intensity levels were analyzed from the original images and graphed using SigmaPlot software and PhotoImpact.
Determination of total intracellular GSH
GSH of various cellular compartments was chemically assayed using the o-phthalaldehyde (OPT)-based method of Hissin et al. 10 Briefly, the cell supernatant was first diluted 10-fold with phosphate-EDTA buffer (pH 8.0). To a tube containing 100 µL of the diluted supernatant, 1.8 mL of phosphate-EDTA buffer and 100 µL of the OPT solution (100 µg) were added. All tubes were then thoroughly mixed and incubated at room temperature (RT) for 15 min. Fluorescence at 420 nm was then determined following activation at 350 nm. FAD-instigated intracellular GSH depletion was further assayed using chloromethyl-fluorescein-diacetate (CMF-DA)-based confocal microscopic imaging technique. Once inside the cell, the esterases cleave off the diacetate group, the resultant chloromethyl group reacts with intracellular thiols exhibiting green fluorescence. Thus, the suppression of fluorescence intensity is an indication of GSH depletion. In this experiment, cells were loaded with 10 µM CMF-DA for 30 min at 37°C to measure the level of intracellular GSH using a Leica SP2 MP Laser Scanning Confocal Microscopy (Leica-Microsystems, Wetzlar, Germany).
siRNA interfering knockdown of survivin and GRP-78
First, survivin expression of HCC Mahlavu cells was ablated using siRNA interfering technique, which was modified as previously described. 11 Briefly, a short oligonucleotide that targets human survivin messenger RNA (mRNA) sequence of 5′-TGGGAGCCAGATGACGACC-3′ and the scrambled siRNA sequence of 5′-AAGGTGGTTGTTTTGTTCACT-3′ was used. The survivin siRNA and scrambled siRNA were inserted into the pSUPERIOR vector to generate pSUPERIOR-survivin-siRNA and pSUPERIOR-scramble-siRNA plasmids, and the insertion was confirmed by DNA sequence analysis. The transfection protocol was carried out by the method previously described.12,13 Briefly, 1.5 × 105 cells were washed twice with PBS and mixed with 0.5 µg of plasmid. We applied 1 pulse of 20 ms under a fixed voltage of 1.4 kV on a pipette-type microporator Neon (Invitrogen Life Technologies, Carlsbad, CA, USA). The stably transfected cells were selected using neomycin for 2 weeks, and the expression of survivin was determined using quantitative reverse transcription real-time polymerase chain reaction (PCR) and western blot analysis. The expression of GRP-78 was knockdowned in Mahlavu cells with siRNA interfering technique as described previously.14,15 Briefly, the target sequence for the human GRP-78 mRNA was 5′-AAGGTTACCCATGCAGTTGTT-3′. The scrambled siRNA sequence was 5′-AAGGTGGTTGTTTTGTTCACT-3′. The GRP-78 siRNA and scrambled siRNA were inserted in the pSUPERIOR vector and transferred into Mahlavu cells. Cells that were successfully transfected into Mahlavu cells were selected by antibiotic resistance as previously described.
Western blot analysis
Protein extract (~40 µg) was separated on 8% or 12% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA) and were blocked in 5% skim milk powder in Tris-buffered saline with Tween 20 (TBST) solution for 1 h at room temperature (RT) followed by incubation with first antibodies against COX-2, GRP-78, Bcl-2, survivin, β-catenin, iNOS, and actin. Following washing with TBST solution, blots were incubated with the appropriate horseradish peroxidase (HRP)-labeled secondary antibody for 1 h at RT. The antigen–antibody complexes were detected by the enhanced chemiluminescence (ECL; Millipore) with a chemiluminescence analyzer.
Statistical analysis
Data are presented as mean ± standard deviation from at least three independent experiments and analyzed using Student’s t-test. A p value of less than 0.05 was considered statistically significant.
Results
FAD triggers ONS via overproduction of ROS/NO concentrating in mitochondrial region
Figure 1 shows that cultivating Hep G2 cells under FAD condition can provoke ONS as evident by the green fluorescence exhibited as the DCF and DAF probes trapped inside the cells were oxidized by ROS that could be detected flowcytometrically (Figure 1(a) and (b)). However, localization of ROS productions using MitoR as an indicator for mitochondria revealed a precise overlapping of DCF and MitoR fluorescences. This result indicated that FAD-triggered ROS production concentrated mainly in the mitochondrial region as detected by confocal microscopy (Figure 1(c)).
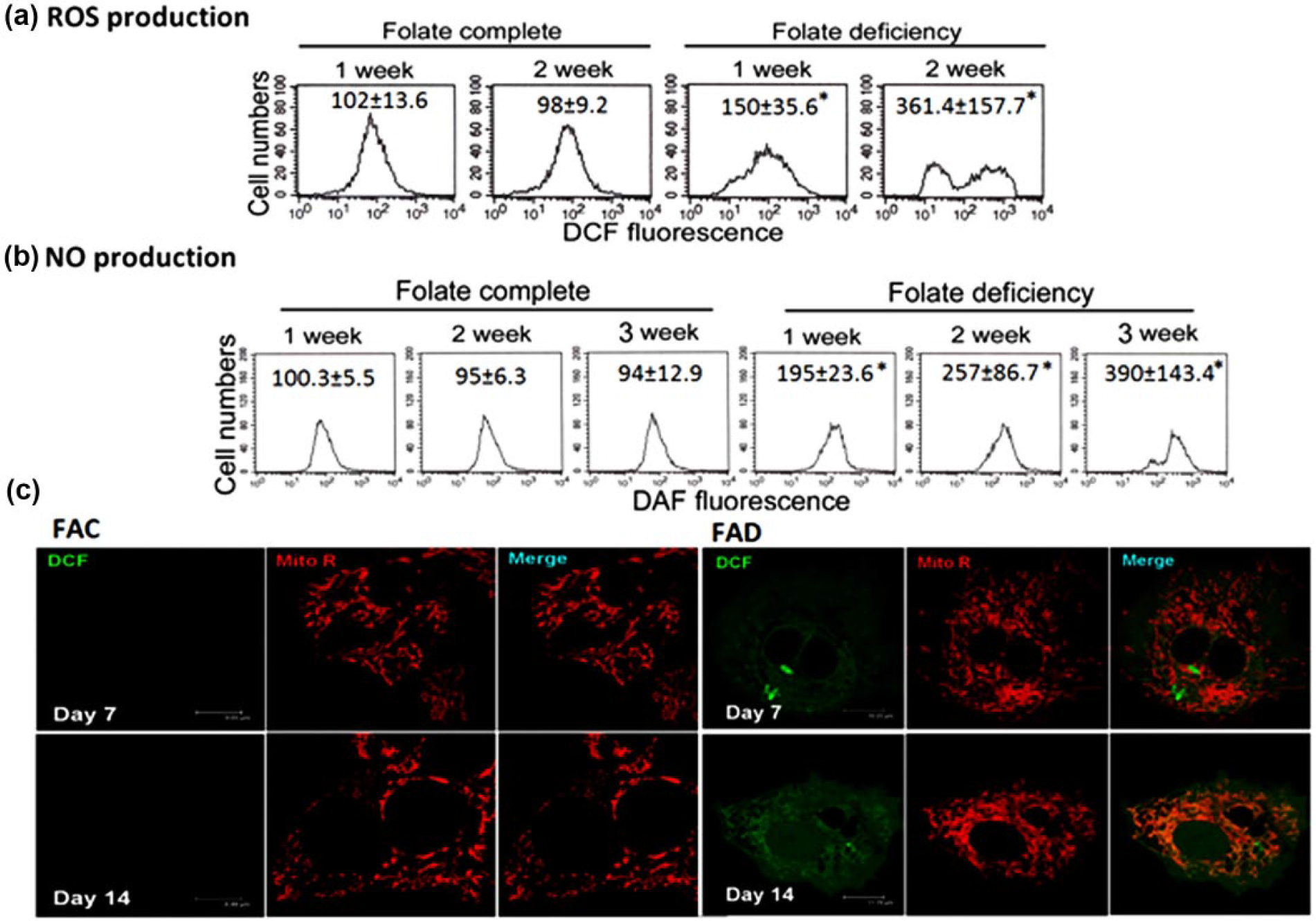
Folate deficiency (FAD) triggers oxidative–nitrosative stress in Hep G2 cells. (a) Production of intracellular ROS in Hep G2 cells cultivated under either FAC or FAD condition was evaluated flowcytometrically using DCF-DA as the probe. At the end of 2-week cultivation period under FAD condition, more than 2-fold increase in ROS production, as reflected by the fluorescence generated when DCF trapped inside the cells was oxidized by ROS, could be detected as compared to FAC control. The values shown are mean ± SD (n = 3 of individual experiments; *p < 0.05 vs control). (b) Production of intracellular NO in Hep G2 cells grown under either FAC or FAD condition was similarly evaluated flowcytometrically using DAF-DA as the probe. At the end of 3-week cultivation period under FAD condition, a 2-fold increase in NO production could be detected as compared to FAC control. Data shown are mean ± SD with at least three independent experiments (*p < 0.05 vs control). (c) Localization of ROS production using Mito R as a probe for mitochondria revealed that cultivation of Hep G2 cells under FAD condition for 2 weeks, a precise overlapping of the fluorescences of DCF and MitoR, could be observed by the confocal microscope. This finding is an indication that FAD-triggered ROS production concentrated mainly in the mitochondrial region.
FAD causes severe downregulation of a pair of anti-apoptotic proteins in Hep G2 cells
During cultivation of Hep G2 cells under FAD condition for 1–2 weeks, severe downregulation of survivin and glucose-regulated protein-78 (GRP-78) could be observed. Coincidentally, β-catenin, an effector regulating Wnt signaling pathway, and COX-2 expressions were concomitantly suppressed when Hep G2 cells were cultured under FAD condition. Furthermore, it is interesting to note that the suppression of β-catenin correlates inversely with the overexpression of E-cadherin (Figure 2(a) and (b)). In addition, the disappearance of pro-caspase 3 indicates the cleavage and activation of caspase 3 leading to apoptosis (Figure 2(d)). Based on these data, the downregulation of survivin during an episode of FAD was ascribable to the disturbance of β-catenin/COX-2/PGE-2 pathway (Figure 2).

FFolate deficiency (FAD) perturbed β-catenin/COX-2/PGE2/Bcl-2 pathway (see Discussion) in Hep G2 cells. At the end of 2-week cultivation under FAD condition, severe downregulation of survivin and GRP-78 as determined by western blot technique (d) correlated excellently with complete disappearance of β-catenin and COX-2 (b and c). Furthermore, the suppression of β-catenin correlated negatively with the expression of E-cadherin (a). It was also interesting to note that the disappearance of pro-caspase 3 band in FAD condition represented that caspase 3-dependent apoptosis had occurred (d).
Novel roles of survivin and GRP-78 as ROS sinker proteins independent of their anti-apoptotic effects
Using a poorly differentiated and a RAP hepatoma Mahlavu cell type with high genetically endowed expression of both survivin and GRP-78 as a study model, we want to address the question that besides their anti-apoptotic attribute, what other functions that these effectors can also bestow. Specifically, we want to find out whether or not they can act as ROS sinkers. Toward this aim, we first knockdowned survivin and GRP-78 separately using siRNA interfering technique followed by growing survivin KD and GRP-78 KD cells under either FAC or FAD conditions. Remarkably, both survivin KD and GRP-78 KD cells could produce extremely high levels of ROS as compared to control cells indicating that both survivin and GRP-78 could act as ROS sinker proteins independent of their anti-apoptotic attributes (Figure 3). Data by western blot also revealed that severe downregulation of antioxidant enzymes such as CAT and GPx might be partially responsible for the survivin KD-mediated overproduction of ROS observed (Figure 4).
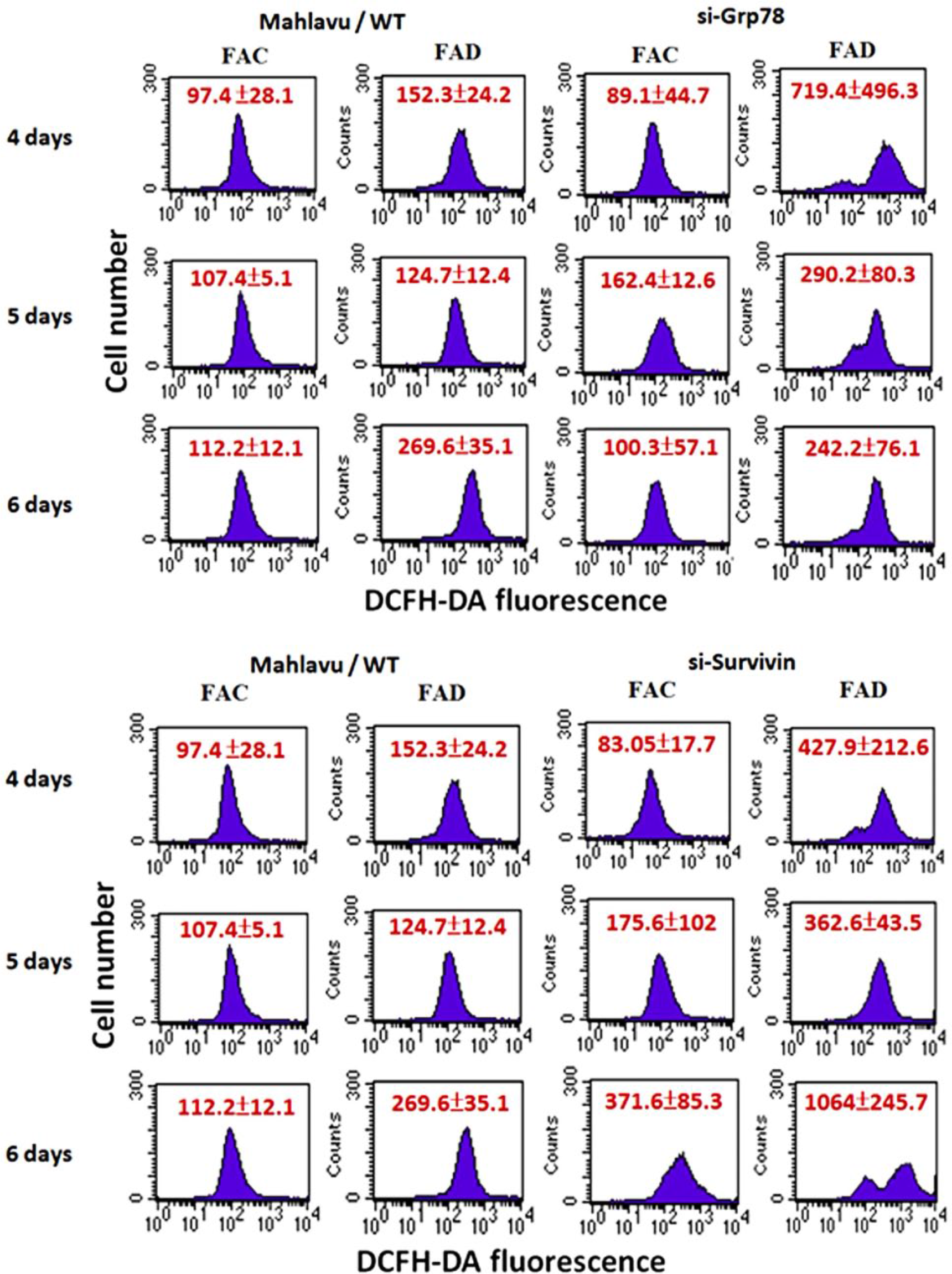
Evidence that survivin and GRP-78 are ROS sinker proteins. Using hepatoma Mahlavu cells which are found to be genetically endowed with strong expression of survivin and GRP-78 as the experimental cell model, we first knockdowned both effectors by siRNA interfering technique followed by growing survivin KD and GRP-78 KD cells under FAC and FAD conditions. Remarkably, both types of KD cells produced extremely high levels of ROS as compared to controls. These data serve as the testimony that both survivin and GRP-78 act as ROS sinkers. Data shown are mean ± SD with at least three independent experiments (*p < 0.05 vs control).

siRNA knockdown of survivin severely downregulates several important ROS-scavenging antioxidant enzymes. We utilized western blot technique to identify that at least three antioxidant enzymes including γ-GCS, catalase (CAT), and glutathione peroxidase (GPx) were severely downregulated in survivin KD cells as compared to the controls. These data clearly implied that survivin KD-mediated overproduction of ROS was at least partially ascribable to the downregulation of important antioxidant enzymes.
FAD could rapidly deplete intracellular GSH contents indiscriminately within various cellular compartments
Using probe-based confocal microscopic detection technique, we demonstrated that culturing Hep G2 cells under FAD condition, the green fluorescence of CMF-GSH complex within the cell was found to be drastically reduced by FAD-induced ROS (Figure 5(a) and (b)). Analysis by software, GSH depletion was found to occur indiscriminately within various cellular compartments. Next, we further confirmed this observation by directly analyzed GSH contents using an established OPT-based fluorometric method. We showed that approximately 70% of GSH contents was found to be depleted at the end of 2-week culture period under FAD condition (Figure 5(c)).

FAD triggers rapid depletion of GSH within various cellular compartments. First, we used probe-based confocal microscopic detection technique to assay GSH depletion instigated by FAD-induced ROS. The probe, CMF-DA, containing a mild thiol reactive group is colorless and nonfluorescent. Once inside the cell, CMF group reacted with GSH, transforming the probe into a cell-impermeant fluorescent dye–thioester adduct. (a) A drastic reduction of CMF-GSH green fluorescence intensity in FAD cell as compared to FAC cell is an indication of GSH depletion phenomenon. (b) GSH was calculated from areas based on a Mito R distribution from the same cell. Intensity values were analyzed per pixal from the cytosolic, the mitochondrial, and the nuclear area of the cells by Leica QWin software. Again a drastic GSH depletion phenomenon could be detected within various cellular compartments. (c) FAD-induced GSH depletion phenomenon was further confirmed by an established fluorometric quantitation of GSH using o-pathalaldehyde reagent. At the end of 2-week cultivation period under FAD condition, approximately 70% of cellular GSH was found to be depleted. The values shown are mean ± SD of three independent experiments (*p < 0.05 vs control).
Insight into the mechanisms associated with FAD-triggered cellular GSH depletion
Three possible processes that may underlie the mechanistic link associated with FAD-induced cellular depletion of GSH can be explored. First, can FAD condition perturb mitochondrial GSH transport system? Bcl-2 has been demonstrated as a novel interacting partner for the 2-oxoglutarate carrier (OGC) and a key regulator of mitochondrial GSH. Thus, we investigate whether or not FAD condition can cause downregulation of Bcl-2 expression in Hep G2 cells. As indicated in Figure 6(a), culturing Hep G2 cells under FAD condition for 2 weeks, severe downregulation of Bcl-2 expression could be observed indicating that GSH transport into mitochondria could be affected. Second, can FAD condition affect cytosolic GSH synthesis? This possibility has also been studied. As indicated in Figure 6(b), Hep G2 cells cultured under FAD condition for 2 weeks caused severe downregulation of γ-glutamylcysteinyl synthetase (γ-GCSh), a catalytic enzyme responsible for the biosynthesis of cytosolic GSH. Thus, reduced GSH synthesis was also partially involved in the FAD-induced GSH depletion. Finally, can FAD condition affect the recycling process of GSH/GSSG in mitochondria? Since this recycling process involved the generation of NADPH via isocitrate dehydrogenase-2 (ICDH-2) that is required for the regeneration of GSSG to GSH catalyzed by GSH reductase. As indicated in Figure 6(c), a severe suppression of ICDH-2 was observed when Hep G2 cells were cultured under FAD condition for 2 weeks indicating that FAD condition could also contribute in GSH depletion process via affecting the regeneration of mitochondrial GSSG back to GSH.
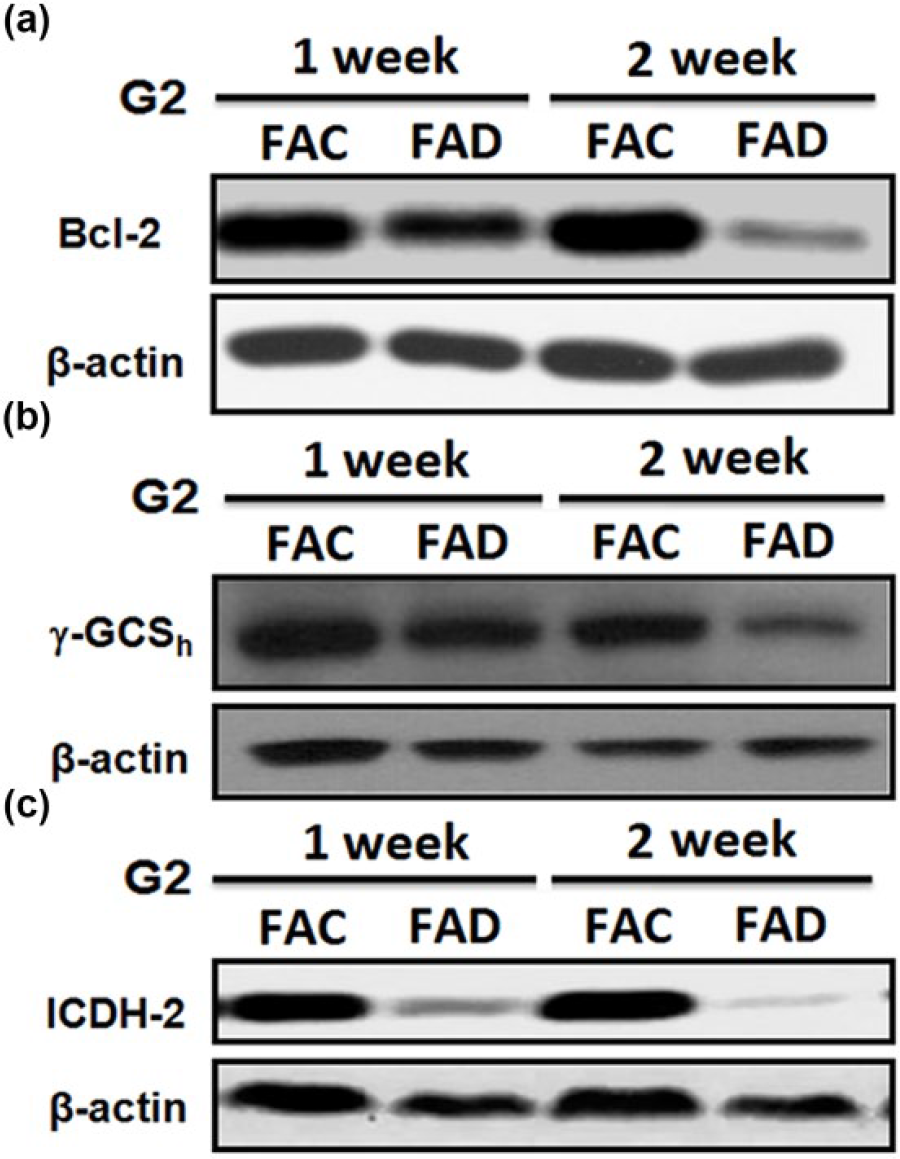
Study on the underlying mechanisms associated with FAD-engendered cellular GSH depletion. (a) FAD condition causes Hep G2 cells to downregulate Bcl-2 expression severely and renders Bcl-2 dependent mitochondrial transporting system abnormal due to the decreased binding of Bcl-2 to its interacting partner 2-oxoglutarate carrier. (b) FAD condition can also cause reduction of cytosolic GSH synthesis via suppression of γ-GCSh, a catalytic enzyme responsible for cytosolic GSH biosynthesis. (c) FAD condition can severely suppress mitochondrial isocitrate dehydrogenase-2 (ICDH-2) which shut down the generation of NADPH necessary for the recycling of oxidized GSH (GSSG) back to reduced GSH catalyzed by mitochondrial GSH reductase. Taken together, these data clearly show that folate plays a pivotal role in the maintenance of redox homeostasis via regulating cytosolic GSH biosynthesis, GSH transporting system, and mitochondrial GSH recycling process.
Discussion
Despite folate has previously been advocated as an antioxidant based on the ability of this micronutrient to scavenge a variety of ROS and non-oxygen-derived free radicals, 6 the information pertaining to whether or not folate can also veritably regulate specific antioxidative pathways other than merely ROS-scavenging activity in vitro has been relatively scanty. In this study, we provide some additional novel results for the first time which strongly attest that this micronutrient can play pivotal role in the upregulation of several prominent antioxidative pathways to meet the justifiable criteria as a genuine antioxidant. First, using Hep G2 cells as the study model, we demonstrated that when these cells were cultured under FAD condition, the expression of two prominent anti-apoptotic effectors including survivin and GRP-78 was found to be profoundly downregulated. These data along with the activation of caspase-3 serve as a testimony that Hep G2 cells have undergone apoptosis. This is a characteristic of HCC RAN phenotypic subclone variant. To further examine the role of GRP-78 and survivin in defending FAD-triggered ONS, we switch our attention to HCC Mahlavu cells, a RAP phenotype capable of overexpressing GRP-78 and survivin constitutively. Silencing of either survivin or GRP-78 in HCC Mahlavu cells with siRNA interfering technique, we further established that both effectors could actually serve as ROS sinkers as reflected by the drastic suppression of the green fluorescence intensity of DCF probe measured flowcytometrically. Interestingly, knockdown of survivin in Mahlavu cells resulted in severe suppression of catalase (CAT) and glutathione peroxidase (GPx) implicating that survivin knockdown–mediated overproduction of ROS might be ascribable, at least in part, to the downregulation of these important antioxidant enzymes (Figure 4).
The canonical Wnt signaling pathway plays a seminal role in the expression of genes including cyclin D, vascular endothelial growth factor (VEGF), and survivin, which involves the translocation of β-catenin into the nucleus.16–20 Using Hep G2 cells as the study model, we found that FAD condition profoundly downregulated survivin, which was shown to correlate with the concomitantly suppressed expression of β-catenin. Along the same vein, our data were also in accordance with the finding reported elsewhere indicating that FAD condition severely inhibited nuclear translocation of β-catenin. 21 In addition, suppressed β-catenin could simultaneously downregulate both in the expression of survivin and GRP-78.9,22 In sharp contrast, when Mahlavu cells, a RAP HCC phenotypic subclone variant, were exposed to FAD condition, the expression of β-catenin was found to be highly upregulated. Concomitantly, E-cadherin was found to be severely downregulated. 23 This phenomenon is ascribable to the promotion of epithelial-to-mesenchymal transition (EMT) evoked by FAD-induced ONS.
Mitochondria are the major intracellular source of ROS. 24 However, maintenance of mitochondrial ROS at physiological levels without oxidative stress is crucial for cell survival. 25 GSH is an endogenous antioxidant and a major player in defending ROS in the mitochondria albeit its content is strictly regulated. However, GSH synthesis occurs exclusively in the cytosol because the required enzymes for its synthesis are absent within mitochondria and the transport of GSH into these organelles is mediated by a specific transporter. Furthermore, the regeneration of oxidized GSSG cannot be carried out in the cytosol because of the retransportation hindrance. Under these premises, three possible processes that may underlie the mechanistic link associated with FAD-induced cellular depletion of GSH can be explored. First, can FAD condition influence mitochondrial GSH transporting system? Apart from its role as a key sentinel for the protection against apoptosis, Bcl-2 is also known to possess a critical antioxidant-like activity owing to its capacity in the regulation of mitochondrial GSH contents.26,27 Thus, mitochondrial GSH is a key endogenous antioxidant and its maintenance is important for cell survival.28,29 Therefore, Bcl-2 is a crucial regulator of the mitochondrial GSH pool through its role as an interacting partner for the 2-OGC. 30 As indicated in Figure 6(a), culturing Hep G2 cells under FAD condition for 2 weeks, severe downregulation of Bcl-2 expression could be observed via perturbing COX-2/PGE-2 pathway indicating that GSH transport into mitochondria could be affected. Consequently, FAD condition could severely deprive mitochondrial GSH. For this reason, the defense mechanism for mitochondria against ROS insults would be lost. Collectively, these data imply that folate can play a pivotal role in the proper maintenance of mitochondrial redox homeostasis of GSH via regulating GSH transporter mediated by Bcl-2.
Next, apart from its role in the regulation of mitochondrial GSH contents via Bcl-2, we addressed the issue of whether or not FAD condition could also affect cytosolic GSH biosynthesis. As indicated in Figure 6(b), Hep G2 cells cultivated under FAD condition for 2 weeks caused severe downregulation of γ-glutamylcysteinyl synthetase heavy chain (γ-GCSh), a catalytic enzyme responsible for the biosynthesis of cytosolic GSH. Thus, reduced GSH synthesis is also partially involved in the FAD-induced GSH depletion. Finally, we addressed the question of whether or not FAD condition could affect the regeneration process of oxidized GSSG back to reduced GSH catalyzed by mitochondrial GSH reductase. Since this regeneration process requires the production of NADPH via ICDH-2 that serves as the cofactor for the regeneration of GSSG back to GSH catalyzed by GSH reductase, we study whether or not FAD condition can affect ICDH-2. As indicated in Figure 5(c), a severe suppression of ICDH-2 was observed when Hep G2 cells were cultivated under FAD condition for 2 weeks. Based on this finding, we thus concluded that FAD condition could also participate in GSH depletion via affecting the recycling of mitochondrial GSSG back to GSH due to the deficiency of NADPH. In other words, FAD condition could perturb at least two important intracellular redox couples including GSH/GSSG system, the dominant redox buffer for most cells,31,32 and NADPH/NADP+ system.33–35
In conclusion, besides being advocated previously by its free-radical-scavenging ability, we now add some novel data to the literature that folate is indeed a genuine and superb antioxidant by the following reasons. First, we uncovered that folate plays a pivotal role in the regulation of the expression of two anti-apoptotic proteins (survivin and GRP-78) which have now been proven by us to act as novel ROS sinkers necessary for the alleviation of oxidative stress. Second, folate can also play a crucial role in the maintenance of mitochondrial redox homeostasis. In this regard, three types of protective mechanisms to alleviate GSH depletion have been delineated. First, folate can maintain the proper function of Bcl-2-dependent mitochondrial GSH transport. Second, folate can properly regulate cytosolic GSH synthesis via upregulating γ-GCSh, a key enzyme responsible for GSH synthesis. Finally, folate plays an important role in the regulation of mitochondrial ICDH-2 which is responsible for providing NADPH necessary for the regeneration of GSSG back to GSH catalyzed by GSH reductase. Taken together, besides being able to plethorically express two prominent ROS sinker proteins, folate can avert cellular GSH depletion through proper maintenance of redox homeostasis via regulating cytosolic GSH biosynthesis, GSH transporting system, and mitochondrial GSH regeneration process.
Footnotes
Acknowledgements
K.-G.L. and C.-F.C. have contributed equally to this article.
Declaration of conflicting interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.