
Abstract
Overexpression of apoptosis-stimulating protein 2 of p53 (ASPP2) induces apoptotic cell death in hepatoma cells (e.g. HepG2 cells) by enhancing the transactivation activity of p53, but long-term ASPP2 overexpression fails to induce more apoptosis since activation of the epidermal growth factor/epidermal growth factor receptor/SOS1 pathway impairs the pro-apoptotic role of ASPP2. In this study, in recombinant adenovirus-ASPP2-infected HepG2 cells, ASPP2 overexpression induces amphiregulin expression in a p53-dependent manner. Although amphiregulin initially contributes to ASPP2-induced apoptosis, it eventually impairs the pro-apoptotic function of ASPP2 by activating the epidermal growth factor/epidermal growth factor receptor/SOS1 pathway, leading to apoptosis resistance. Moreover, blocking soluble amphiregulin with a neutralizing antibody also significantly increased apoptotic cell death of HepG2 cells due to treatment with methyl methanesulfonate, cisplatin, or a recombinant p53 adenovirus, suggesting that the function of amphiregulin involved in inhibiting apoptosis may be a common mechanism by which hepatoma cells escape from stimulus-induced apoptosis. Thus, our data elucidate an apoptosis-evasion mechanism in hepatocellular carcinoma and have potential implications for hepatocellular carcinoma therapy.
Introduction
Apoptosis is involved in the regulation of many physiological and pathological processes in cells. 1 Insufficient cell death by apoptosis promotes tumorigenesis. 2 Moreover, evasion of apoptosis is one of the hallmarks of tumors that result in treatment resistance. 2 The discovery of apoptotic escape mechanisms has great potential for designing novel therapeutic strategies for human cancer. 2 Hepatocellular carcinoma (HCC) represents one of the most difficult cancers to treat. 3 Up to now, the current therapeutic strategies designed to induce apoptosis are still not effective enough to completely eliminate HCC. 4
Apoptosis-stimulating protein 2 of p53 (ASPP2) is characterized by the presence of ankyrin repeats, an SH3 domain, and a protein-rich region. 5 ASPP2 binds to p53 through its C-terminus to stimulate the transactivation function of p53 on the promoters of pro-apoptotic genes. 5 Other studies also demonstrated that ASPP2 can induce apoptosis in a p53-independent manner. 6 Thus, it is likely that ASPP2 overexpression can be used to treat cancer by inducing apoptotic cell death, and our previous results indicate that induction of ASPP2 overexpression can promote apoptotic cell death in hepatoma cells, which emphasizes the value of ASPP2 in treating HCC.7,8 However, when ASPP2 overexpression is prolonged, hepatoma cells will develop a mechanism of apoptotic resistance, which leads to a failure to induce more apoptosis by ASPP2. 7 The activation of the epidermal growth factor (EGF)/epidermal growth factor receptor (EGFR)/son of sevenless 1 (SOS1) pathway contributes to apoptosis resistance in hepatoma cells with prolonged ASPP2 overexpression because activation of this pathway impairs the binding of ASPP2-p53 complexes to the promoters of pro-apoptotic genes and inhibits the expression of pro-apoptotic genes. 7 However, how the EGF/EGFR/SOS1 pathway is activated is still unclear.
Amphiregulin (AREG) is a member of the ligand family that binds to EGFR. The binding of AREG to EGFR activates intracellular signaling cascades that promote cell survival, proliferation, and motility. 9 In various neoplasms, including colon, lung, breast, prostate, and liver cancer, AREG is involved in promoting tumor growth, and its expression levels are always elevated. 10 A recent study demonstrated that p53 binds to and transactivates the AREG promoter, which induces AREG expression. 11 Because ASPP2 can enhance p53 transactivation ability, it is rational to hypothesize whether ASPP2 overexpression will induce AREG expression and whether AREG will lead to cell survival by impairing the pro-apoptotic function of ASPP2.
Here, we identified that AREG produced by HepG2 cells that overexpress ASPP2 activates the EGF/EGFR/SOS1 pathway, which results in apoptosis resistance in hepatoma cells and leads to cell survival.
Materials and methods
Cell culture and treatment
Wild-type HepG2 cells and HepG2 cells with stable p53 knockdown (HepG2-p53-kd) were grown in Dulbecco’s modified Eagle’s medium (DMEM). DMEM was supplemented with 10% fetal bovine serum (FBS). The HepG2-p53-kd cell line was kindly provided by Dr. James Ou (Molecular Microbiology and Immunology, Keck School of Medicine of USC). Cells were infected with recombinant adenovirus (rAd)-ASPP2 for 24, 48, and 72 h. Lentiviral vectors that express human AREG shRNA (AREG-sh) and control shRNA (Ctrl-sh) were purchased from Novus Biologicals. A total of 5 × 105 HepG2 cells were seeded in a 6-cm plate and infected with a lentiviral dose of 2.5 × 106 the following day. Puromycin (2 µg/ml) was used to select HepG2 cells that stably expressed the Ctrl-sh and AREG-sh. Neutralizing antibody of AREG (ne-AREG) (eBioscience) was used to block the function of soluble AREG. The cells were grown on glass cover slips for immunofluorescence studies.
Enzyme-linked immunosorbent assay
At each time point, supernatant was collected and stored at −80°C. The levels of soluble AREG and EGF in the supernatant were detected by enzyme-linked immunosorbent assay (ELISA) kits (Abcam) according to the manufacturer’s protocol.
Real-time polymerase chain reaction
The RNeasy Mini Kit (Qiagen) was used to isolate total RNA from the cultured cells. Reverse transcription was used to synthesize first strand complementary DNA (cDNA) using the SuperScript II First-Strand Synthesis System for reverse transcription polymerase chain reaction (RT-PCR; Invitrogen). The SYBR Green method was used to detect the double-stranded DNA (dsDNA) products during the real-time PCR reaction. The messenger RNA (mRNA) content was normalized to the expression of the housekeeping gene β-actin. The following specific primer sequences were used for real-time PCR: β-actin, 5′-GCCCTGAGGCACTCTTCCA-3′ (forward) and 5′-CGGATGTCCACGTCACACTT-3′ (reverse). The AREG primers were obtained from the PrimePCR TM SYBR Green® Assay (Bio-Rad).
Immunoblot assay
Cell lysates were subjected to immunoblot analysis as previously described. 7 Briefly, total cell lysates were separated on 10% or 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels, and the separated proteins were transferred to polyvinylidene fluoride (PVDF) membranes. The protein blots were blocked with 5% non-fat milk and sequentially probed with specific primary antibodies and horseradish-peroxidase-conjugated secondary antibodies. The detection of specific proteins on the blots was achieved using an enhanced chemiluminescence system (Pierce), and the results were captured on either X-ray film or ImageQuant LAS 4000. Densitometric analysis was performed using the Image-Pro Plus analysis software. The antibodies for AREG and EGFR were purchased from Abcam. Anti-SOS1 and β-actin antibodies were acquired from Santa Cruz Biotechnology.
Chromatin immunoprecipitation
PCR of the AREG promoter sequence was performed using 35 cycles as follows: 94°C for 15 s and 65°C for 45 s. The resulting PCR products were analyzed on 1.5% tris–acetate–ethylenediaminetetraacetic acid (EDTA) (TAE)-agarose gels. 12 The AREG-specific primer sequences are as follows: 5′-GTACTTTTACATCTAAATACGGA-3′ and 5′-GTGCGTAAGGATTCGCTGAGAGGAA-3′. 11
Cell death and apoptosis analysis
Cell death was quantified with the calcein acetoxymethyl (AM) ester/propidium iodide (PI) assay (Thermo) as previously described. 13 Briefly, calcein-AM and PI were added into the supernatant, and the cells were incubated with calcein-AM/PI for 15 min. Apoptosis was detected using the terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay (Promega) as previously described. 13 Briefly, the cells were fixed with 10% paraformaldehyde/phosphate-buffered saline (PBS) for 15 min and then incubated in 1% Triton X-100/PBS for 5 min. A TUNEL detection solution was dropped onto the glass cover slips, and the cells were incubated at 37°C in the dark for 60 min. Nuclear staining with 4′,6-diamidino-2-phenylindole (DAPI) was conducted for 5 min after rinsing with PBS. Finally, the slips were mounted with 50% glycerol after rinsing with PBS. The images of the calcein-AM/PI and TUNEL assays were obtained by fluorescence microscopy. A quantitative cell death/apoptosis analysis was performed by counting more than 1000 cells in each examination.
Luciferase reporter assay
The detailed methods for the luciferase reporter assay have been previously described. 12 Briefly, the PCR product of the AREG promoter was cloned into the pGL3-basic luciferase plasmid (Promega) using standard techniques. Cells were transfected with the AREG reporter plasmid for 8, 16, 24, 48, and 72 h. Relative light unit values were normalized to a β-galactosidase signal. One microgram of each indicated AREG promoter-reporter plasmid and pRSV β-galactosidase plasmid was used for all transfections.
Statistical analysis
All data shown are the results of at least three independent experiments and are expressed as the mean ± standard deviation (SD). The differences between groups were compared using Student’s t-test. Differences were considered statistically significant at confidence levels of p < 0.05, p < 0.01, and p < 0.001 as indicated.
Results
ASPP2 overexpression induces AREG expression in HepG2 cells
HepG2 cells were infected by rAd-ASPP2 for 8, 16, 24, 48, and 72 h. Real-time PCR assay showed increased mRNA levels of AREG at 16 and 24 h (Figure 1(a)). In HepG2 cells transfected with the AREG luciferase reporter, ASPP2 overexpression also dramatically increased AREG luciferase activity at 16 and 24 h, suggesting that ASPP2 can induce the transcription of AREG (Figure 1(b)). Immunoblotting and ELISA assays further showed that ASPP2 overexpression increased the levels of intracellular and soluble AREG, respectively (Figure 1(c) and (d)).
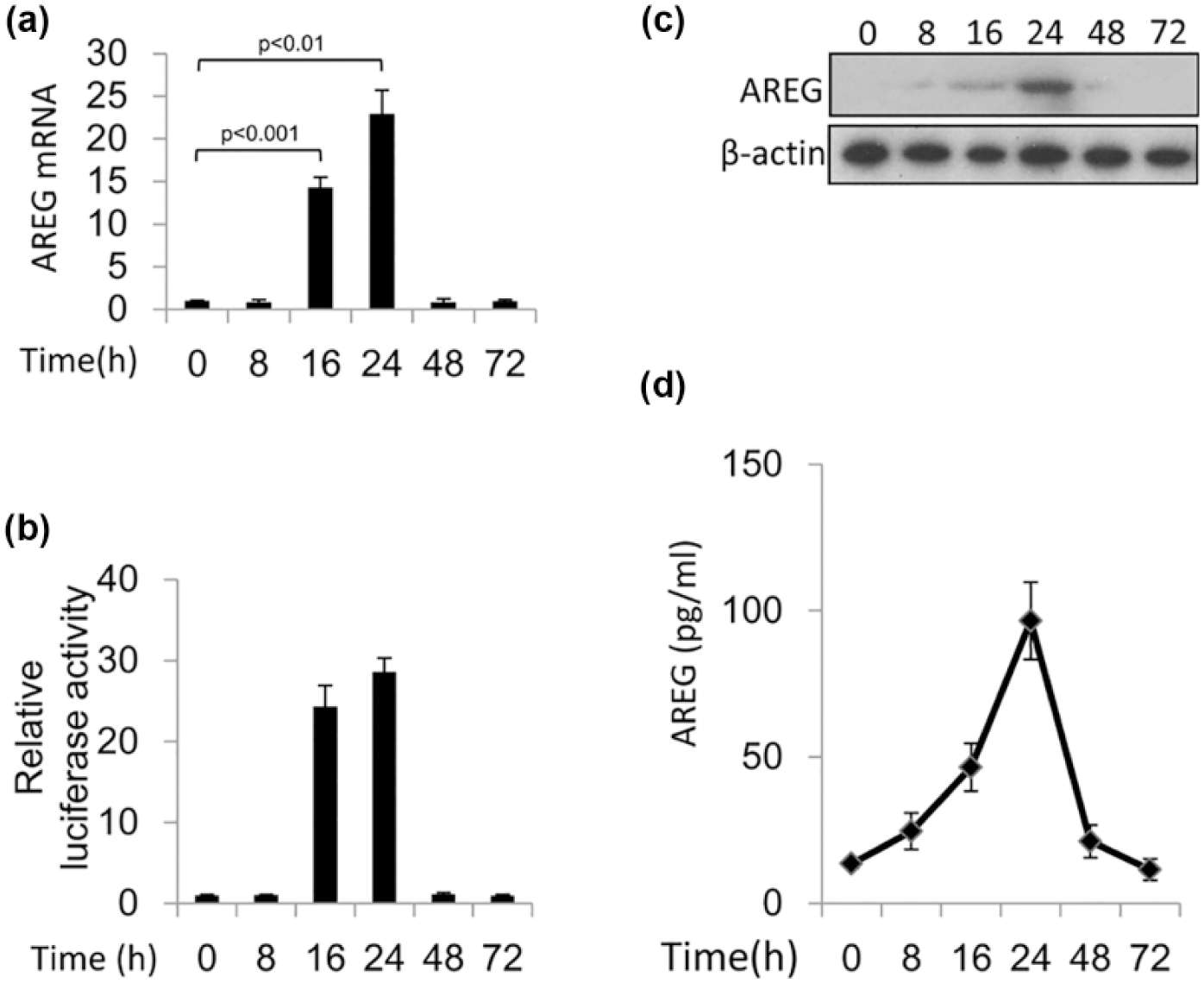
ASPP2 overexpression induces AREG expression: (a and c) HepG2 cells were infected by rAd-ASPP2 for 8, 16, 24, 48, and 72 h. (a) Real-time PCR and immunoblot assays were used to detect the expression of AREG. (b) Luciferase activity of the indicated pGL3-AREG promoter-reporter constructs after transfection into HepG2 cells is shown. (d) HepG2 cells were infected by rAd-ASPP2 for 8, 16, 24, 48, and 72 h.
ASPP2 induces AREG expression by binding to its promoter in a p53-dependent manner
p53 is reported to induce AREG expression by directly binding to its promoter, and p53-induced AREG plays a pro-apoptotic function by impairing the anti-apoptotic function of Bcl-2. 11 Here, the binding of p53 to the AREG promoter was identified in HepG2 cells after rAd-ASPP2 infection for 24 h but not at 48 or 72 h (Figure 2(a)). The binding of ASPP2 to the AREG promoter was also identified in HepG2 cells infected with rAd-ASPP2 for 24 h (Figure 2(b)); however, at this time point, the binding of ASPP2 to the AREG promoter could not be identified in HepG2 cells with stable knockdown of p53 (Figure 2(c)). These data suggest that ASPP2 binds to the AREG promoter in a p53-dependent manner.

ASPP2 binds to the AREG promoter in a p53-dependent manner. The chromatin immunoprecipitation assays are shown. (a and b) Wild-type HepG2 cells and (c) HepG2 cells with stable knockdown of p53 were infected with rAd-ASPP2 for the indicated duration. (a) Anti-p53 and (b and c) anti-ASPP2 antibodies were used for chromatin immunoprecipitation, followed by PCR and gel electrophoresis in ethidium bromide–stained agarose.
AREG is involved in ASPP2 overexpression–induced apoptosis at 24 h but impairs subsequent apoptotic cell death at 48 and 72 h
Here, knockdown of AREG via shRNA reduced ASPP2 overexpression–induced apoptosis and cell death at 24 h, suggesting that AREG plays a pro-apoptotic role at this time point (Figure 3(a) and (b)). Apoptosis and cell death could not be induced at 48 and 72 h in HepG2 cells after rAd-ASPP2 infection; however, in HepG2 cells with stable knockdown of AREG, ASPP2 overexpression–induced apoptosis and cell death at these two time points (Figure 3(a) and (b)). Moreover, in HepG2 cells with stable knockdown of AREG, ASPP2 overexpression–induced apoptosis and cell death at 48 and 72 h were higher than those at 24 h (Figure 3(a) and (b)). These data suggest that AREG initially plays a pro-apoptotic role, but it will eventually impair the pro-apoptotic function of ASPP2.
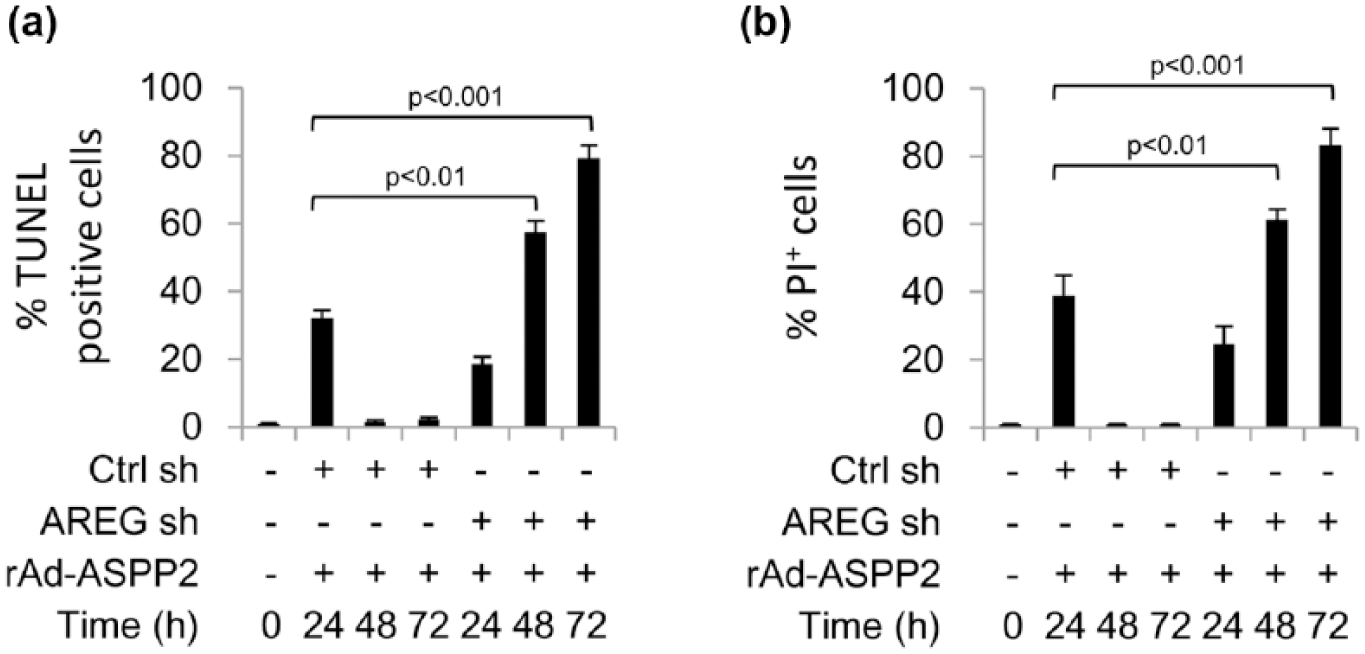
AREG is involved in initial ASPP2 overexpression–induced apoptotic cell death but eventually impairs the pro-apoptotic role of ASPP2. AREG shRNA and control shRNA were used to produce a stable knockdown of AREG in HepG2 cells (AREG-sh) and a control cell line (Ctrl-sh), respectively. rAd-ASPP2 was used to infect the two cell lines (AREG-sh and Ctrl-sh) for the indicated durations. (a) TUNEL and (b) calcein-AM/PI assays were used to detect apoptosis and cell death, respectively.
Soluble AREG-activated EGF/EGFR/SOS1 pathway impairs the pro-apoptotic function of ASPP2
Our previous study has demonstrated that the activation of the EGF/EGFR/SOS1 pathway provides a survival signal for HepG2 cells after infection with rAd-ASPP2 for 48 and 72 h. 7 Here, ASPP2 overexpression enhanced EGF levels in the supernatant at 48 and 72 h, and knockdown of AREG significantly reduced the levels of soluble EGF at these two time points (Figure 4(a)). Induction of ASPP2 overexpression can increase the levels of EGF and EGFR, and then the binding of EGF to EGFR induces the expression of SOS1 at 48 and 72 h, which impairs the pro-apoptotic function of ASPP2. 7 Here, in HepG2 cells with stable knockdown of AREG, ASPP2 overexpression did not induce the expression of EGFR and SOS1 (Figure 4(b)). These data suggest that AREG is required for the expression of EGF, EGFR, and SOS1.

Soluble AREG-activated EGF/EGFR/SOS1 pathway impairs the pro-apoptotic function of ASPP2. (a) ELISA detection of the EGF levels in the supernatant of HepG2 cells with stable knockdown of AREG (AREG-sh) with or without rAd-ASPP2 infection for the indicated durations. Ctrl-sh is a control cell line. (b) Immunoblot detection of the expression of AREG, EGFR, and SOS1 in AREG-sh and Ctrl-sh cells after rAd-ASPP2 infection for the indicated durations. (c) HepG2 cells were infected with rAd-ASPP2 for 6 h and co-cultured (upper layer) with normal HepG2 cells (lower layer) for 48 h with or without neutralizing antibody of AREG (ne-AREG) added into the co-culture system. Then, rAd-ASPP2 was used to infect HepG2 cells on the surface of lower layer for 24 and 48 h, and TUNEL staining was used to detect apoptosis in HepG2 cells on the surface of the lower layer.
Moreover, we used a transwell system to separately culture rAd-ASPP2-infected HepG2 cells (upper layer) and uninfected HepG2 cells (lower layer) for 48 h; rAd-ASPP2 was then used to infect HepG2 cells on the surface of the lower layer for 24 and 48 h. Interestingly, rAd-ASPP2 infection could not induce apoptosis in the HepG2 cells in the lower layer at either time point (Figure 4(c)). However, when a neutralizing antibody of AREG (ne-AREG) was added into the co-culture system, rAd-ASPP2 infection induced apoptosis in HepG2 cells on the surface of the lower layer, suggesting that the soluble AREG produced by rAd-ASPP2-infected HepG2 cells (upper layer) provides a survival signal for the HepG2 cells on the lower layer to resist ASPP2-induced apoptosis (Figure 4(c)).
AREG contributes to an anti-apoptotic function in hepatoma cells in response to many signals for cell death
To detect whether induction of AREG is a common mechanism by which hepatoma cells develop resistance to apoptosis, we used methyl methanesulfonate (MMS), cisplatin, and rAd-p53-induced p53 overexpression as different death signals. In the transwell system, HepG2 cells on the upper layer treated with MMS (Figure 5(a)), cisplatin (Figure 5(b)), or rAd-p53 infection (Figure 5(c)) were co-cultured with HepG2 cells without any treatment (lower layer). We identified that when the function of soluble AREG was blocked by ne-AREG, treatment with MMS (Figure 5(a)), cisplatin (Figure 5(b)), or rAd-p53 infection (Figure 5(c)) induced more apoptosis in HepG2 cells on the lower layer. Thus, at least in our experimental setting, the induction of soluble AREG is a common method for contributing to the production of an anti-apoptotic signal in the remaining hepatoma cells in response to a death stimulus.

Soluble AREG also inhibits apoptosis induced by the other pro-apoptotic signals. HepG2 cells were treated with (a) MMS, (b) cisplatin, or (c) rAd-p53 infection with or without pretreatment with the neutralizing antibody of AREG.
Discussion
Escape from apoptosis contributes to drug resistance in many tumor cells, which always leads to the failure of chemotherapeutics. Here, we identified that AREG contributes to apoptosis escape in hepatoma cells stimulated with apoptotic signals. AREG is a ligand of EGFR and is synthesized as a membrane-anchored precursor protein that can engage in juxtacrine signaling on adjacent cells. 14 AREG is secreted and behaves either as an autocrine or paracrine factor, and TACE/ADAM17 are the main proteases that are involved in the proteolytic processing of AREG. 15 The binding of AREG to EGFR can activate some intracellular signaling cascades that govern cell survival, proliferation, and motility. 10 Physiologically, AREG can affect the development and maturation of mammary glands, bone tissue, and oocytes; however, chronic elevation of AREG expression is always associated with inflammatory and/or neoplastic development. 9 Here, we identified that the function of intracellular AREG is associated with the induction of apoptosis, but soluble AREG is an anti-apoptotic factor, which might explain why AREG could play a dual role in the control of cell survival, proliferation, and death.
AREG is synthesized as a type II transmembrane protein (pro-AREG). Both the generation of soluble AREG and AREG ectodomain shedding are dependent on the proteolytic activity of the transmembrane proteinase ADAM-17/TACE. 15 In particular, the release of AREG can be induced by many signals, including cytokines, UV radiation, and chemotherapeutic drugs such as 5-fluorouracil. 16 In our study, the release of AREG from apoptotic HepG2 cells after rAd-ASPP2 infection provided a survival signal for the remaining cells by activating the EGF/EGFR2/SOS1 pathway.
Currently, the number of reports that describe the role of AREG in tumorigenesis and cancer cell biology is overwhelming. In many types of cancer, including HCC, the expression of AREG is upregulated. 10 Some studies support a pro-oncogenic role of AREG in several tumor types. 17 For example, AREG overexpression provides self-sufficient growth and survival signals in liver cancer. 18 In one model where AREG involvement in inflammation-driven hepatocarcinogenesis was reported, AREG overexpression in severely inflamed livers contributed to liver fibrogenesis and finally led to liver cancer development by establishing an autocrine loop. 19 Thus, detecting the expression of AREG is beneficial as an early sign of liver cancer development.
In fact, AREG is also regarded as a critical factor for the induction of resistance to chemotherapeutic agents in liver cancer. 18 A large number of studies have shown that AREG contributes to cisplatin, doxorubicin, and sorafenib resistance in HCC. One study has shown that an EGFR inhibitor and AREG neutralization could re-establish the function of sorafenib in inducing cell death in HCC. 20 Our study further demonstrates that the AREG-activated EGF/EGFR2/SOS1 pathway leads to resistance to pro-apoptotic signals. We believe that our results are helpful to identify novel targets and to improve the therapeutic efficacy in the treatment of liver cancer.
Footnotes
Acknowledgements
Kai Liu and Dongdong Lin contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by National Natural Science Foundation of China (81402556, 81272266), International Cooperation and Exchanges NSFC (81361120401), Foundation of Qingdao Municipal Science and Technology Commission (14-6-1-6-zdzx-12), Talent Project from Beijing Health System (2013-3-074), and the capital health research and development of special Foundation (2014-1-1151).