
Abstract
Multicomponent molecular modifications such as DNA methylation may offer sensitive and specific cervical intraepithelial neoplasia and cervical cancer biomarkers. In this study, we tested cervical tissues at various stages of tumor progression for 5-methylcytosine and 5-hydroxymethylcytosine levels and also DNA promoter methylation profile of a panel of genes for its diagnostic potential. In total, 5-methylcytosine, 5-hydroxymethylcytosine, and promoter methylation of 33 genes were evaluated by reversed-phase high-performance liquid chromatography, enzyme-linked immunosorbent assay based technique, and bisulfate-based next generation sequencing. The 5-methylcytosine and 5-hydroxymethylcytosine contents were significantly reduced in squamous cell carcinoma and receiver operating characteristic curve analysis showed a significant difference in (1) 5-methylcytosine between normal and squamous cell carcinoma tissues (area under the curve = 0.946) and (2) 5-hydroxymethylcytosine levels among normal, squamous intraepithelial lesions and squamous cell carcinoma. Analyses of our next generation sequencing results and data from five independent published studies consisting of 191 normal, 10 low-grade squamous intraepithelial lesions, 21 high-grade squamous intraepithelial lesions, and 335 malignant tissues identified a panel of nine genes (ARHGAP6, DAPK1, HAND2, NKX2-2, NNAT, PCDH10, PROX1, PITX2, and RAB6C) which could effectively discriminate among the various groups with sensitivity and specificity of 80%–100% (p < 0.05). Furthermore, 12 gene promoters (ARHGAP6, HAND2, LHX9, HEY2, NKX2-2, PCDH10, PITX2, PROX1, TBX3, IKBKG, RAB6C, and DAPK1) were also methylated in one or more of the cervical cancer cell lines tested. The global and gene-specific methylation of the panel of genes identified in our study may serve as useful biomarkers for the early detection and clinical management of cervical cancer.
Introduction
Cervical cancer is one of the major causes of cancer-related deaths among women worldwide and is strongly associated with high-risk human papillomavirus (HPV) infection. Although early cytomorphological screening and vaccines against HPV are anticipated to reduce the cervical cancer burden, 1 it is projected that by the year 2020, there will be 42% increase in cervical cancer incidence globally (http://globocan.iarc.fr/Default.aspx). In India, cervical cancer alone accounted for 24% (62,416) of all cancer cases in women who succumbed to the disease in the period 2015–2016. Epigenetic mechanisms leading to global hypomethylation and gene-specific hypermethylation play a key role in carcinogenesis, including cervical cancer.2,3 Removal of methyl group from the cytosine residue of the DNA resulting in global hypomethylation is mediated by several non-enzymatic mechanisms or by the action of ten–eleven translocation (TET) family of alpha-glutarate-dependent oxygenases. 4 Oxidative by-products of 5-hydroxymethylcytosine (5-hmC), such as 5-formylcytosine (5-fC) and 5-carboxylcytosine (5-caC), are part of cytosine deaminase pathway and participate in DNA repair process. 5 Recently, role of 5-fC and 5-caC in affecting the rate of polymerase II nucleotide incorporation, thereby influencing the rate of transcription, is also reported. 6 It is now understood that “cytosine codes” (various modifications of cytosine such as 5-mC, 5-hmC, 5-fC or 5-caC) govern epigenetic plasticity that may regulate gene expression.
The aberrant methylation of genes has been reported as an early and frequent event during cervical carcinogenesis. 7 In addition to their role in transcription silencing and tumor development, the hypermethylation of promoters at specific CpG sites can be sensitive and specific diagnostic and prognostic markers.8,9 For example, DNA promoter methylation of glutathione S-transferase pi 1 (GSTP1) and O-6-methylguanine-DNA methyltransferase (MGMT1) is reported as a potential diagnostic marker for glioblastoma and prostate cancer.10–12 It has been shown that 95% of the adenocarcinoma of the prostate have methylated GSTP1 promoter, while the normal tissues were apparently negative. 13 The MGMT1 DNA methylation at specific CpG sites is shown to be an important prognostic marker for gliobastoma and response to treatment. 10 The diagnostic and prognostic significance of methylated genes in cervical cancer have been extensively reviewed.2,14,15 A majority of the cervical cancer DNA methylation studies have validated genes which are also reported in other cancers, and only a few of these markers are specific for cervical cancer with high sensitivity and specificity. 16 In addition, studies have implicated ethnicity as one of the confounding factors for the development of DNA methylation–based biomarkers. For example, cyclin-dependent kinase inhibitor 2A (p16INK4a) gene is hypermethylated in individuals with hepatocellular carcinoma (HCC) from Japan and is unmethylated in individuals from Taiwan.17,18
Although DNA methylation changes represent one of the stable and earliest changes during carcinogenesis, efforts are needed to discover and validate these findings using population-based studies. There is also a need for the novel targets useful for diagnostics, prognostics, and therapeutic interventions, which can be used uniformly across the population. In this study, we have identified new DNA methylation markers with high sensitivity and specificity for cervical cancer screening. Furthermore, we have tested the global 5-methylcytosine (5-mC) and 5-hmC levels, evaluated the methylation status of large number of gene promoters in a panel of tissues representing progressive stages of cervical cancer, and performed in silico validation in independent datasets.
Materials and methods
Cell line maintenance and patient cohort
Three cervical cancer cell lines (SiHa, HeLa, and CaSki) were purchased from American Type Culture Collection (ATCC, USA) and subcultured according to the standard protocol (http://www.atcc.org/). Samples of the human subjects who visited either for screening or for the diagnosis or treatment of cervical cancer were obtained from hospitals affiliated to the Kasturba Medical College (KMC) (Manipal and Mangalore, Karnataka, India). In total, three lymphoblastoid cell lines (LCLs) were generated in our laboratory by infecting peripheral blood mononuclear cells (PBMCs) with Epstein–Barr virus (EBV), as published earlier. 19 In brief, PBMCs were separated from 10 mL of blood by Ficoll-Hypaque lymphocyte separation medium (GE Healthcare, USA), according to manufacturer’s instructions, and infected with EBV isolated from marmoset B-cell line (B95-8), as published earlier. 19 The transformed cell lines were cultured in RPMI containing 10% fetal bovine serum (FBS), confirmed by light microscopy and immunophenotyping using CD23, CD58, and CD19 antibodies.
Individuals who were already on the treatment or those who had synchronous cancers were excluded from the study. Samples were collected after obtaining the written informed consent from the participants in accordance with ethics committee of Kasturba Hospital (KH) Manipal, Manipal University. The samples were histologically reviewed by pathologists and classified according to the Bethesda system. 20 Histopathologically confirmed premalignant and squamous cell carcinoma (SCC) cells that contained more than 80% of abnormal and tumor cells were used for DNA isolation. The global 5-mC levels were estimated in the DNA of 20 normal, 35 SCC, 3 cervical cancer cell lines (SiHa, HeLa, and CaSki), and 3 LCLs. The 5-hmC content was estimated in 10 each of normal, squamous intraepithelial lesions (SILs), SCC, and 3 cervical cancer cell lines (SiHa, HeLa, and CaSki). For next generation sequencing (NGS)-based bisulfite genomic sequencing, 20 each of normal, SIL, SCC, and 3 cervical cancer cell lines were used.
DNA extraction and bisulfite conversion
DNA was isolated by digesting the cells/tissues with proteinase K followed by standard phenol chloroform and ethanol precipitation method, while bisulfite modification was performed using EZ DNA Methylation Kit (Zymo Research, USA). Bisulfite-converted DNA (100 ng) was amplified using forward and reverse primers under the following conditions: 95°C for 5 min; 34 cycles of 95°C for 30 s, respective annealing temperature for 45 s, 72°C for 1 min; and a final extension of 72°C for 5 min (Table 1). The polymerase chain reaction (PCR) products were gel purified and used for NGS.21,22
The list of primers used in this study.

F: forward primer; R: reverse primer; TSS: Transcription start site.
Methylation analysis by NGS
The bisulfite PCR product was purified and used for library construction according to Ion Xpress™ Plus Fragment Library Kit (Thermo Fisher Scientific, USA). In brief, PCR products of eight genes/samples were pooled at a final concentration of 25 nM in 100 µL and were fragmented using Ion Shear™ Plus Reagents according to manufacturer’s protocol (Thermo Fisher Scientific, USA). The fragmented DNAs were purified using Agencourt AMPure XP beads (Beckman Coulter, USA), ligated with Ion P1 adapter, and individual samples were barcoded with Ion Xpress barcode. The adapter-ligated, nick-translated, repaired DNA was purified using Agencourt AMPure XP beads. Approximately, 330 bp target library was prepared from unamplified library (200 base read length) using E-Gel SizeSelect™ 2% Agarose Gel (Thermo Fisher Scientific, USA). The size selected product (200 base read with target length of 330 bp) was then amplified and purified using Ion Xpress™ Plus Fragment Library Kit. The quality and quantity of the amplified library were analyzed by High Sensitivity DNA kit using Bioanalyzer (Agilent Technologies, USA) and Qubit® dsDNA HS Assay Kit using Qubit fluorometer (Thermo Fisher Scientific, USA). Each pooled library (100 pM) was subjected to emulsion PCR, and enrichment was carried out using Ion PGM™ Hi-Q™ OT2 reagent in Ion OneTouch™ 2 system (Thermo Fisher Scientific, USA). The templates for sequencing were enriched by Ion OneTouch™ ES and sequenced using 316v2 Chips in the Ion Personal Genome Machine (PGM) Sequencer (Thermo Fisher Scientific, USA).
Analysis of NGS data
We have used the following pipeline to analyze bisulfite sequencing raw reads. In brief, the raw reads were analyzed for the sequence quality using FastQC. Nucleotides with quality scores <20 were trimmed from the ends using FASTX-Toolkit (version 0.0.14; http://hannonlab.cshl.edu/fastx_toolkit/). After mapping the sequences to the genomic regions, it was subsequently analyzed using NextGenMap 0.4.12. 23 Sequences were also aligned against converted and unconverted reference genomic regions to analyze the extent of bisulfite conversion. The degree of methylation at each CpG site was estimated using BiQAnalyzer HT software (http://biq-analyzer-ht.bioinf.mpi-inf.mpg.de/). 24
HPV DNA typing
The HPV genotyping was performed, as published earlier, with appropriate positive and negative controls.25,26 The HPV strains were identified by National Center for Biotechnology Information’s Basic Local Alignment Search Tool (NCBIBLAST) search and HPV database (https://pave.niaid.nih.gov/). HPV negative samples were scored as negative after at least two independent rounds of testing.
Global methylation analysis by reversed-phase high-performance liquid chromatography
Global 5-mC levels were estimated using reversed-phase high-performance liquid chromatography (RP-HPLC) as described earlier.27–29 Briefly, 2 µg of genomic DNA was digested with DNase1 (Ambion, USA), denatured at 100°C for 5 min, chilled on ice, and incubated with 2 U each of Nuclease P1 (Sigma, USA) and calf intestinal phosphatase (New England BioLabs, USA) according to manufacturer’s instruction. The sample was passed through Quia quick spin column (Qiagen, USA), lyophilized, and subjected to chromatographic separation (10 µL) under ambient column temperature, maintaining the samples at 4°C using isocratic conditions with a flow rate of 1 mL/min in HPLC instrument (Waters, USA), consisting of dipotassium hydrogen phosphate buffer (K2HPO4, pH 4.1) supplemented with 10% methanol and C-18 Grace Vydac smart column at 260 and 280 nm. Standards such as dC, 5-dmC, dT, dA, and dG were used to identify the retention time of these nucleosides. Peaks corresponding to 5-mC and other nucleosides were monitored over 30 min interval. The relative degree of methylation in the DNA samples was measured using the following formula: % of 5-mdC = A5mdC / (A5mdC + AdC) × 100.
Global 5-hmC estimation
The global 5-hmc was estimated using MethylFlash™ Hydroxymethylated DNA Quantification Kit (EpiGentek, USA) according to manufacturer’s instruction. The 5-hmC was estimated in 200 ng of DNA using the capture and detection antibodies followed by colorimetric detection of absorbance at 450 nm using Varioskan™ Flash Multimode Reader (Thermo Fisher Scientific, USA). The amount of hydroxymethylated DNA is calculated using the following formula according to the manufacturer’s instruction

where S is the amount of input sample DNA in nanograms; P is the amount of input positive control (HC5) in nanograms; 5* is a factor to normalize 5-hmC in the positive control to 100%, as the positive control contains only 20% of 5-hmC; HC4 is the negative control which is a methylated polynucleotide containing 20% of 5-mC. The HC5 positive control is a hydroxymethylated polynucleotide containing 20% of 5-hmC.
Public datasets
Independent validation of our findings was performed using cervical cancer methylation and gene expression microarray datasets, available at ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) and the Cancer Genome Atlas (TCGA; https://tcga-data.nci.nih.gov/tcga/) using cancer browser (https://genome-cancer.ucsc.edu) or cBioPortal (www.cbioportal.org). A total of four independent datasets were created consisting of 557 microarrays for methylation and 66 microarrays for gene expression. The methylation dataset consisted of 191 normal, 10 low-grade SILs (LSILs), 21 high-grade SILs (HSILs), and 335 tumor samples, while the gene expression dataset consisted of 24 normal, 33 tumor, and 3 cervical cancer cell lines (Supplementary Table S1). DNA methylation and expression analysis were also performed using Methylation and Expression Database of Normal and Tumor Tissues (MENT; http://mgrc.kribb.re.kr:8080/MENT/), a database of DNA Methylation and gene expression in Human Cancer (MethHC; http://methhc.mbc.nctu.edu.tw/), Gene Expression across of Normal and Tumor Tissues (GENT; http://medical-genome.kribb.re.kr/GENT/), and Wanderer (http://maplab.imppc.org/wanderer/) databases. HumanMethylation27 and HumanMethylation450 BeadChip data were extracted from ArrayExpress (E-GEOD-30760, E-GEOD-30759, E-GEOD-41384, and E-GEOD-46306 datasets) database. The clinical information and detailed description of the dataset are available at ArrayExpress. The cervical SCC and endocervical adenocarcinoma (CESC) dataset consisting of DNA methylation data (n = 260; Illumina Infinium Human methylation 450 K bead array, Illumina, USA) and RNA-seq data (n = 256) were available at the time of our analysis. A β value ≥0.2 was considered as methylated. The β value was considered as significantly hypermethylated only if the value was found in more than 5% of tumors, as published earlier. 30
Bioinformatics and statistical analysis
Circos and CRAN Package “pheatmap” (https://cran.r-project.org/web/packages/pheatmap/index.html) were used for circular representation of the significantly differentially methylated regions and heat maps, respectively. 31 Results of all the assays were shown as mean ± standard deviation (SD). Student’s t test, analysis of variance (ANOVA), Kruskal–Wallis test, and Fisher’s exact test were performed using Microsoft Excel (Microsoft, USA) and GraphPad Prism 5 (GraphPad Software, USA). A p value ≤ 0.05 was considered as statistically significant. The sensitivity and specificity analysis were performed by MedCalc (http://www.medcalc.org/calc/diagnostic_test.php) and GraphPad Prism 5.
For random forest (RF) analysis, we have averaged the methylation value of all the CpG sites within the given region of each gene for each sample. For RF analysis, methylation was considered as an independent variable, tumor condition was considered a dependent variable, and 1000 random trees were built using bootstrap sampling as the tree building algorithm. Herein, software used 63.2% of randomly selected samples as training set and remaining ones as validation cohort. Following this, misclassification error (OOB error) was calculated by confusion matrix. RF and area under the curve (AUC) were calculated using randomForest and ROCR using R package, respectively.32–36
Results
Clinical characteristics of patients
This study consisted of 75 participants (20 normal, 20 SIL consisting of 6 LSIL and 14 HSIL, and 35 SCC samples). In our samples, we have combined both LSILs and HSILs together as SILs. The median age of normal, SIL, and SCC samples was 46, 53, and 58 years, respectively. All women were sexually active and non-smokers. Two in the normal group and one in the malignant group were nulliparous. Except for three women in the normal group, none had undergone a Pap test before. The HPV prevalence in normal, SIL, and tumor samples was found to be 20%, 65%, and 90%, respectively.
Cervical cancer genome is globally hypomethylated
Global DNA methylation at the cytosine residues was estimated in normal (n = 20), SCC (n = 35), three cervical cancer cell lines (SiHa, HeLa, and CaSki), and three LCLs by RP-HPLC. There was a significant reduction in global methylation content between normal and tumor samples (p < 0.0001; Figure 1(a)–(c)). The 5-mC content was significantly lower in cervical cancer cell lines (SiHa = 1.8%, HeLa = 1.9%, and CaSKi = 2.1%; Figure 1(d)). However, the 5-mC levels in LCLs were higher than the cervical cancer cell lines (3.84 vs 1.93; Figure 1(d)). The receiver operating characteristic (ROC) curve analysis showed a significant difference in 5-mC content between normal and tumor samples with an AUC = 0.946 and p < 0.0001 (Figure 1(e)). Taken together, our result shows that the cervical cancer genome is globally hypomethylated and can be useful for discriminating tumor from normal tissues.

Cervical cancer genome shows reduced 5-mC and 5-hmC level. The 5-mC content was estimated by RP-HPLC in 20 normal, 35 SCC, 3 cervical cancer cell lines (SiHa, HeLa, and CaSki), and 3 LCLs. (a and b) The HPLC profile from normal and tumor samples. (c) The 5-mC levels in 20 normal and 35 SCC samples. The mean 5-mC content in normal and SCC samples was significantly different (normal = 3.5 vs SCC = 2.6) analyzed by t test (***p < 0.0001). (d) The 5-mC estimation in cervical cancer cell lines and LCLs. The 5-mC levels were also found to significantly lower in cervical cancer cell lines on comparison with LCLs (1.93 vs 3.84). (e) The diagnostic potential of 5-mC to discriminate SCC from normal samples was performed using ROC curve analysis. The AUC was 0.94 with p < 0.0001. (f) The 5-hmC estimation in 10 each of normal, SIL, and SCC, and 3 cervical cancer cell lines (SiHa, HeLa, and CaSki), respectively. The mean 5-hmC level in normal, SIL, and SCC samples were 0.40, 0.16, and 0.08, respectively. The 5-hmC levels were significantly lower in SIL and SCC samples when compared with normal samples as well as between SIL and SCC samples (p < 0.05). The mean fold difference between SIL versus normal, tumor versus normal, and SCC versus SIL was 2.5-, 5.0-, and 2.0-folds, respectively (p < 0.05). The 5-hmC level in cervical cancer cell lines, namely SiHa, HeLa, and CaSki was 0.017, 0.007, and 0.022, respectively. (g) The ROC analysis of 5-hmC in normal versus SIL, normal versus SCC, and SIL versus SCC, respectively. The AUC was 0.79, 0.92, and 0.63 in normal versus SIL, normal versus SCC, and SIL versus SCC, respectively.
Decreased 5-hmC levels in SIL and tumor sample
The 5-hmC levels were estimated in 10 each of normal, SIL, SCC, and cell lines. The mean 5-hmC levels in normal, SIL, and SCC samples were 0.40, 0.16, and 0.08, respectively (Figure 1(f)). The 5-hmC levels were significantly lower in SIL and SCC when compared to normal samples as well as between SIL and SCC samples (p < 0.05). The mean fold difference between various groups (SIL vs normal, SCC vs normal, and tumor vs SIL) was 2.5-, 5.0-, and 2.0-folds, respectively (p < 0.05). The 5-hmC levels in SiHa, HeLa, and CaSKi cervical cancer cell lines were 0.017, 0.007, and 0.022, respectively (Figure 1(f)). The AUC of 5-hmC between normal and SIL, normal and tumor, and SIL and tumor tissues were 0.79, 0.92, and 0.64, respectively (Figure 1(g)).
Methylation analysis in cervical cancer cell lines
We have analyzed the methylation status of 33 gene promoters in SiHa, CaSki, and HeLa cell lines and compared against the gene expression data from Gene Expression Omnibus (GEO) and GENT databases (Table 2). A total of 14 gene promoters were identified as methylated in at least one of the three cell lines tested (Table 2). Gene expression levels in normal, tumor, and cervical cancer cell lines were analyzed using the Affymetrix expression array data from NCBI (GSE9750 dataset) and GENT database (Table 2). Among the 33 genes tested, 21 of them showed significantly differential expression between normal and tumor tissues (p < 0.05; Table 2). Among the 15 methylated genes tested, 11 of them showed inverse relationship between methylation levels and gene expression.
DNA methylation and its correlation with gene expression.
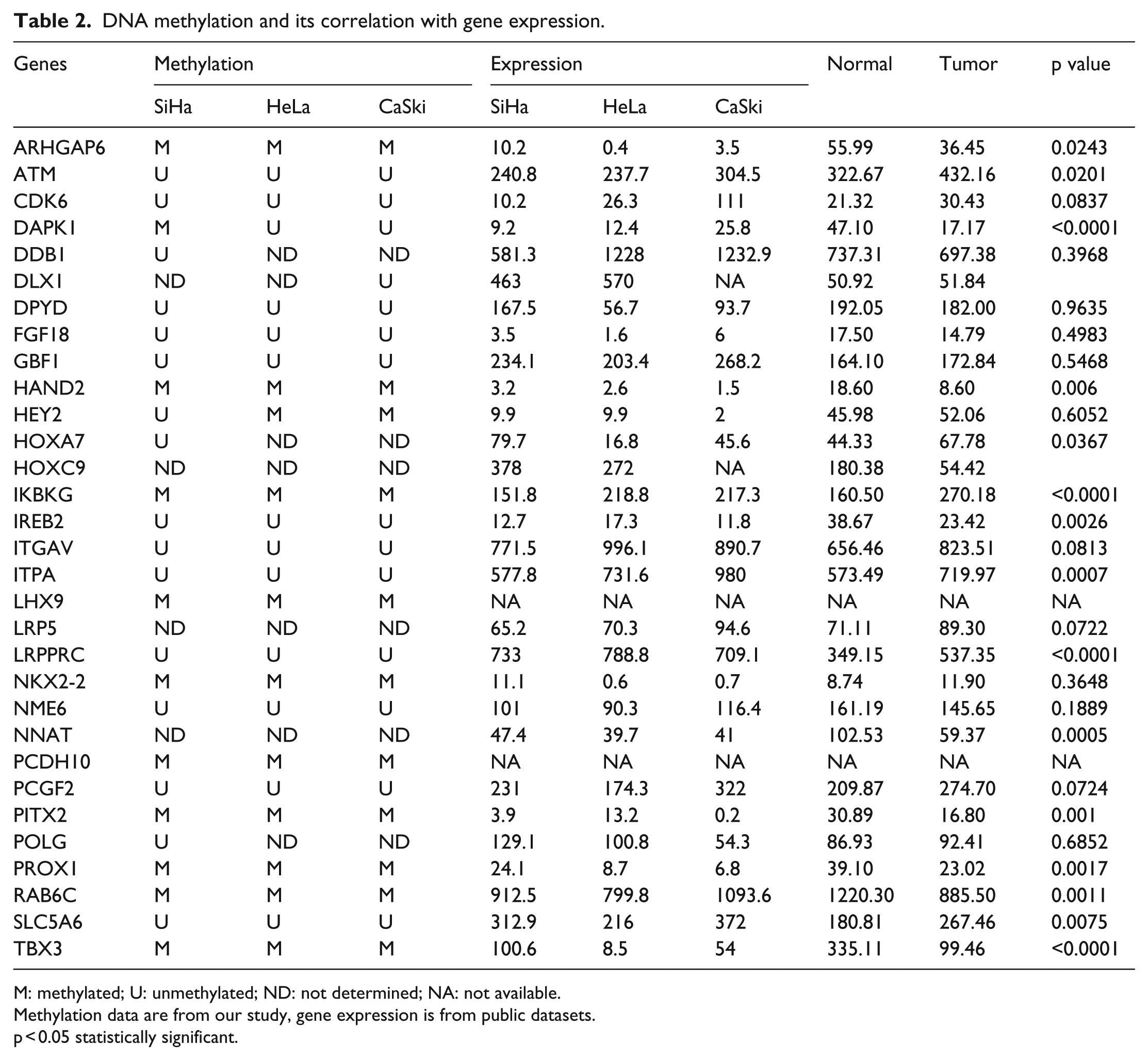
M: methylated; U: unmethylated; ND: not determined; NA: not available.
Methylation data are from our study, gene expression is from public datasets.
p < 0.05 statistically significant.
DNA promoter methylation analysis in cervical samples
Initial screening of 33 gene promoters in normal, SIL, and SCC tissues identified 8 genes as differentially and significantly methylated. These were subsequently screened in another set of 10 samples belonging to each category. Among the 33 gene promoters tested, 8 of them (ARHGAP6, DAPK1 (death-associated protein kinase 1), HAND2 (heart and neural crest derivatives expressed 2), NKX2-2 (NK2 homeobox 2), PCDH10 (Protocadherin 10), PITX2 (paired-like homeodomain 2), PROX1, and RAB6C) showed significant difference in methylation either between normal and SCC or between SIL and SCC tissues (Figures 2 and 3; Table 3). The frequency of DNA methylation in all the 33 genes is shown in Supplementary Table S2. Furthermore, except DAPK1, remaining seven genes showed a significant potential to discriminate tumor from normal samples or tumor from SIL samples (Table 3). However, DAPK1 was found to discriminate only normal tissues from SCC. Furthermore, all the eight genes tested did not discriminate the normal from SIL samples. LHX9 (LIM homeobox 9), IKBKG, and TBX3 (T-box protein 3) were commonly methylated in normal, SIL, and SCC tissues, while ITPA, HOXC9, HEY2, and HOXA7 were methylated in very low percentage of SCC tissues. The gene promoters such as ATM (ataxia-telangiectasia mutated), BTG2, CDK6, DDB1, DPYD, FGF18 (fibroblast growth factor 18), GBF1, IREB2, LRP5, PCGF2, and POLG were unmethylated in all the categories of samples tested. When the individual CpG sites of eight significantly differentially methylated genes were analyzed, several of them showed significant hypermethylation among normal versus tumor or SIL versus SCC tissues (Supplementary Tables S2 and S3).

The methylation analysis of eight differentially methylated gene promoters identified in our study groups. The methylation analysis was performed in 20 each of normal, SIL, and SCC samples by NGS-based bisulfite genomic sequencing. The box plot represents methylation level averaged over all CpG sites analyzed for each categories of samples. The line graph represents the methylation levels averaged over each CpG site of individual categories of samples. Asterisks (*) indicate statistical significance as calculated by ANOVA and Kruskal–Wallis test (p value ≤ 0.05 was considered as statistically significant). The significantly differentially methylated individual CpG sites are shown in Supplementary Table S3.

Heat map of eight differentially methylated genes in 20 each of normal, SIL, and SCC samples, and 3 cervical cancer cell lines (SiHa, HeLa, and CaSki). Samples arranged as normal, SIL, SCC, and SiHa; HeLa and CaSki (horizontal); and CpG sites (vertical). The frequency of methylation is represented by colors with blue and red representing 0%–100% methylation. The heat map was drawn using R package (CRAN) of pheatmap tool.
The list of eight significantly differentially methylated genes and their diagnostic significance.

U: unmethylated; M: methylated; SIL: squamous intraepithelial lesion; AUC: area under the curve; SCC: squamous cell carcinoma.
DNA promoter methylation analysis of public datasets
We have analyzed the methylation status of 33 genes in four public datasets. In Dataset 1 (E-GEOD-30760), promoter CpG sites belonging to six (ARHGAP6, DAPK1, HAND2, NKX2-2, NNAT (Neuronatin), and PITX2) and three (SLC5A6, ITPA, and ITGAV) genes were hypermethylated and hypomethylated, respectively, in tumor samples (153 normal vs 62 tumors). In Dataset 2 (E-GEOD-30759), consisting of 15 normal and 48 tumor samples, six gene promoters such as ARHGAP6, DAPK1, HAND2, NKX2-2, NNAT, and PITX2 were significantly hypermethylated in tumor tissues. However, DLX1, HOXA7, HOXC9, LHX9, PROX1, and TBX3 gene promoters were also hypermethylated in less than 50% of the samples analyzed. In Dataset 3 (E-GEOD-41384), seven gene promoters (DAPK1, DLX1, HOXA7, LRPPRC, NNAT, PITX2, and SLC5A6) were hypermethylated in LSIL samples while eight gene promoters (ARHGAP6, DAPK1, HAND2, HOXA7, NKX2-2, NNAT, PITX2, and PROX1) were hypermethylated in HSIL when compared with normal tissues. The promoters of ARHGAP6, DAPK1, DLX1, HAND2, HOXA7, FGF18, LRP5, NKX2-2, NNAT, PITX2, and TBX3 were significantly hypermethylated in tumors when compared to the normal tissues. In Dataset 4 (E-GEOD-46306), which consisted of 20 normal, 18 HSIL, and 6 tumor samples, DAPK1, NNAT, and SLC5A6 gene promoters were hypermethylated in HSILs, while ARHGAP6, DAPK1, FGF18, HAND2, NNAT, and PITX2 were hypermethylated in tumors when compared with normal tissues. Although there was a difference in the extent of methylation in each study for the same gene, the pattern of methylation was consistent when compared to the normal tissues (Supplementary Table S4).
Sensitivity, specificity, and diagnostic utility of methylation markers
In order to identify the diagnostic significance and robustness of the markers, sensitivity and specificity analysis was performed. First, we investigated the diagnostic significance of methylation markers in our samples followed by independent validation in four public datasets. In our samples and among the eight significantly differentially methylated genes, the sensitivity of PROX1, PCDH10, and ARHGAP6 was 50%, 60%, and 80%, respectively, while the specificity was 95%, 100%, and 100%, respectively, for distinguishing SIL from normal tissues. For normal versus tumor samples, ARHGAP6, DAPK1, HAND2, NKX2-2, PCDH10, PITX2, PROX1, and RAB6C showed sensitivity of 65%–95% and specificity of 60%–100% (p < 0.05). For SIL versus tumor tissues, five genes (DAPK1, PITX2, PROX1, HAND2, and NKX2-2) showed sensitivity of 65%, 90%, 90%, 95%, and 95% and specificity of 90%, 65%, 50%, 75%, and 65%, respectively (p < 0.05; Figure 4; Table 3). The diagnostic potential of genes identified as aberrantly methylated in public datasets is summarized in Supplementary Tables and Figures.

(a) Circular representation of the eight significantly differentially methylated genes. In total, 33 genes were profiled in 20 each of normal, SIL, SCC, and 3 cervical cancer cell lines (SiHa, HeLa, and CaSki) by NGS-based bisulfite genomic sequencing. The circle represents the eight significantly differentially methylated genes. Blue, yellow, and red represent the normal, SIL, and SCC samples. The percentage indicates the extent of methylation. (b–d) The ROC curve analysis of average methylation of the eight genes. The AUC was 0.65, 0.93, and 0.87 for normal versus SIL, normal versus SCC, and SIL versus SCC, respectively (p value < 0.05 was considered as statistically significant).
Discussion
The aim of this study was to investigate the diagnostic relevance of (1) global cytosine methylation, (2) global 5-hmC, and (3) promoter DNA methylation profile of a panel of genes in SIL and SCC. We have also compared our results with the published large datasets from other geographical regions and ethnicity. This is probably the first report to show the clinical utility of 5-mC, 5-hmC, and DNA methylation in a panel of genes in a single study. By ROC curve and sensitivity and specificity analyses, we have identified a panel of genes with potential for clinical management of cervical cancer. Cytosine methylation is an important and widely studied epigenetic modification that is frequently altered in diverse cancers.37–39 Global hypomethylation and gene-specific hypermethylation are the most common and frequent epigenetic modifications known to regulate diverse aspects of tumorigenesis.37,38 Although methylation in mammals is mostly seen at 5′ of cytosine at CpG dinucleotide context, there are also reports of less frequent non-CpG cytosine methylation. 40 During carcinogenesis, both global loss of DNA methylation and gene-specific hypermethylation of anticancer genes have been reported in different types of cancer, including cervical cancer.2,37–39
Global loss of DNA methylation is primarily seen in repetitive elements, comprising approximately half of the human genome. Besides, progressive loss of DNA methylation may lead to activation of proto-oncogenes and transposable elements resulting in genomic instability and thus contributing to carcinogenesis. 41 Similar to other published studies, we have identified significant loss of 5-mC content in cervical cancer tissues and cell lines compared to normal tissues.42,43 Discovery of 5-hmC was considered as “sixth base” and is formed as an oxidative product of 5-mC by a highly regulated enzymatic process controlled by TET family (TET1, TET2, and TET3) of proteins. 4 The distribution of 5-hmC is reported in many different tissues and cell types, and is abundant in embryonic stem cells and adult neuronal cells. 44 Although the biological function of 5-hmC is yet to be completely understood, many studies have reported the global loss of 5-hmC in cancer45–47 and this may be related to reduced 5-mc levels. More recently, existence of 5-hmC inside mitochondria has also been reported. 48 The ROC curve analysis of 5-hmC levels was significant in distinguishing all the pertinent tissue categories tested. A recent study showed that global reduction of 5-hmC in cervical carcinoma is correlated with International Federation of Gynecology and Obstetrics (FIGO) stage, histologic grade, metastatic lymph node, and vascular invasion. 49 Furthermore, the reduced 5-hmC levels were associated with significant decrease in overall survival rate of the patients. 49 Taken together, our study along with other reports suggest that the global loss of 5-hmC during cervical carcinogenesis has clinical relevance. Further study needs to be performed in a large number of samples to elaborate these conclusions.
In our study, we have used an unbiased approach to select the candidate gene promoters to discover their diagnostic significance in cervical cancer. This was accomplished first by identification and validation of panel of gene promoters followed by confirmation of their significance from independent studies consisting of five large panels of datasets. Combined analysis of our study and public datasets identified ARHGAP6, DAPK1, HAND2, PROX1, PCDH10, NKX2-2, NNAT, RAB6C, and PITX2 gene promoters with more than 80% sensitivity and specificity to discriminate between normal, premalignant, or tumor tissues. Moreover, some of the gene promoters (DAPK1, HEY2, LRPPRC, PCDH10, PROX1, RAB6C, and RBBP8) showed more than 80% sensitivity to discriminate normal, premalignant, and malignant tissues but these were not considered due to their lower specificity.
Among the 33 gene promoters screened, PROX1, PCDH10, and RAB6C showed very high sensitivity (80%–100%) and specificity (70%–100%) in distinguishing normal versus premalignant tissues: PROX1, PCDH10, DAPK1, HAND2, PITX2, and NKX2-2 (sensitivity: 80%–100% and specificity: 80%–100%; p < 0.05) for normal versus tumor tissues; and DAPK1, HAND2, and NKX2-2 (sensitivity: 70%–90% and specificity: 80%–90%; p < 0.05) for premalignant versus tumor tissues, respectively. The results of this study are promising and encouraging to deploy in the clinical practice for cervical cancer diagnosis. When these gene promoters were mapped using publicly available independent studies, we were able to identify a consistent pattern of methylation for selected CpG sites. In addition, comparison of these gene promoters in public datasets allowed us to identify additional genes with high sensitivity and specificity. Gene promoters such as ARHGAP6, HAND2, DAPK1, NNAT, NKX2-2, and PITX2 exhibited remarkably high sensitivity and specificity of >90% in distinguishing one among the three categories. Although several gene promoters validated in our dataset were significantly hypermethylated when compared to normal tissues, they were inconsistent in public datasets due to low methylation status and lower sensitivity and specificity. For example, while PROX1 showed very high sensitivity and specificity in our samples, these were absent in public datasets. However, NNAT promoter showed very high sensitivity and specificity in the public datasets and was less sensitive in our samples. This may be attributed to different methods employed, region of the gene used for analysis, and ethnic variations among the population tested. Based on our results, we propose that the hypermethylation of ARHGAP6, DAPK1, HAND2, NKX2-2, NNAT, PROX1, PCDH10, PITX2, and RAB6C makes them potential targets, as they were significantly methylated in more than two datasets.
Several studies have used high throughput microarray or NGS technologies to identify the differentially methylated signatures of clinical relevance.50–52 Despite the differences in the method used for discovery, ethnic differences among the populations, and other confounding factors, the studies have still identified common methylation patterns among the identified genes.53,54 Our study has identified co-methylation and co-expression of multiple genes affecting the same pathway. Moreover, the gene ontology and pathway analysis identified functionally and biologically relevant processes and pathways in cancer.
Among the 33 gene promoters screened, only 3 of them (NNAT, PCDH10, and DAPK1) were previously reported as hypermethylated in cervical cancer, while rest of the 30 gene promoters were found to be novel targets for cervical cancer.55,56 One of the limitations of this study is the moderate clinical sample size with limited samples in the premalignant group. In India, unlike in Western countries, routine screening is not popular, and therefore, few women are diagnosed at the stage of premalignant stage. Moreover, samples which did not contain more than 80% of diseased cells were excluded from the study. Some of the genes identified in our analysis were novel and showed high sensitivity and specificity to cervical cancer progression. However, their mechanism of action and role in cervical cancer progression need to be explored.
Rho GTPase–activating protein 6 (ARHGAP6), a member of rhoGAP family is involved in actin polymerization and cell motility. ARHGAP6 is aberrantly expressed in colorectal cancer and might serve as a biomarker for colorectal cancer. 57 ATM protein belongs to PI3/PI4–kinase family and is an important cell cycle–checkpoint kinase reported as hypermethylated in human colorectal cancer cell lines, oral SCCs, breast cancers, and brain tumors.58–61 In addition to this, methylation of ATM promoter is correlated with radiosensitivity in colorectal cancers. 59 FGF18 is a fibroblast growth factor family member possessing cell survival and mitogenic activities and is involved in cell growth, morphogenesis, tumor growth, and invasion by regulation of actin cytoskeleton, PI3K, and fibroblast growth factor receptor (FGFR) signaling. FGF18 is downregulated in response to demethylating agent treatment in bladder cancer. 62 HAND2 encodes transcription factors belonging to basic helix-loop-helix (bHLH) family and is downregulated in endometrial cancer by promoter hypermethylation. 63 It is essential for cardiac morphogenesis, development, and regulation of angiogenesis and is a part of nuclear factor of activated T cells (NFAT) and cardiac hypertrophy signaling pathway. Hypermethylation of HAND2 is also reported in lung cancer. 64 Hes-related family bHLH transcription factor with YRPW motif 2 (HEY2), a transcription factor belonging to the bHLH-type family, acts as a downstream effector of Notch signaling. 65 The downregulation of HOXA7, belonging to the homeobox group of transcription factors and essential for embryonic development, is reported to be associated with leukemia. The hypermethylation of HOXA7 is reported in lung carcinoma and is associated with aggressive behavior in meningiomas.39,66
LHX9, a developmentally expressed transcription factor, participates in the development of bi-potential gonads in mice and humans. 67 The aberrant methylation and silencing of LHX9 is reported in childhood malignant gliomas. 68 NKX2-2 is a homeobox-containing gene involved in the morphogenesis of central nervous system (CNS) and is required for maintenance of NEUROD1 expression in pancreas. 69 PCDH10, belonging to the protocadherin family, is a potential calcium-dependent cell adhesion molecule associated with Dravet syndrome. It inhibits cancer cell migration and is hypermethylated in cervical and gastric cancers.55,70 Hypermethylation and reduced expression of PCDH10 show poor survival in gastric cancer, metastasis in colorectal cancer, and overall poor survival in bladder cancer. 71 PITX2, belonging to the RIEG/PITX homeobox family, is involved in control of cell proliferation and morphogenesis.72,73 PITX2 is associated with Wnt and transforming growth factor (TGF)-beta signaling and is hypermethylated in breast, prostate, and may other cancers. Hypermethylation of PITX2 is associated with poor survival in breast cancer, and it is a prognostic marker of biochemical recurrence in prostate cancer.74,75 Propero homeobox 1 (PROX1) is a homeobox transcription factor and is associated with breast angiosarcoma and kaposiform hemangioendothelioma. 76 PROX1 is a tumor suppressor gene often hypermethylated in hematological malignancies, sporadic breast cancer, and oral cancer.77,78 PROX1 expression in oral cancer has shown to reduce cell proliferation. 79
TBX3 is involved in the regulation of developmental process and is associated with ulnar–mammary syndrome and holt–oram syndrome. 80 It is hypermethylated in bladder cancer and acts as pTa-specific prognostic marker. 81 TBX3 overexpression is also reported in a number of cancers. In melanoma, TBX3 overexpression is associated with promotion and invasion of melanoma, while in breast cancer, it is shown to interact with histone deacetylases (HDACs).82,83 NNAT, an imprinted gene, is involved in the regulation of ion channels during brain development, and hypermethylation of NNAT locus occurs frequently in pediatric acute leukemia. 84 RAB6C, a member of RAS oncogene family, regulates centrosome duplication and cell cycle progression by guanosine triphosphate (GTP) binding and GTPase activity. RAB6C is hypermethylated and downregulated in drug resistant breast cancer cell lines and is known to inhibit p53 regulated expression. 85 DAPK1 is a calcium/calmodulin-dependent serine/threonine kinase which inhibits cell survival and triggers apoptosis and autophagy. Hypermethylation leading to transcriptional silencing of DAPK1 and reactivation by demethylating agents is reported in many cancers.56,86,87
Conclusion
In conclusion, we have shown that during cervical carcinogenesis, the genome undergoes significant reduction in 5-mC and 5-hmC content. We have also shown that estimation of 5-mC or 5-hmC or profiling for promoter methylation of a panel of genes (ARHGAP6, DAPK1, HAND2, NKX2-2, NNAT, PROX1, PCDH10, PITX2, and RAB6C) may act as a potential tool for early detection and screening of cervical cancer. Furthermore, these methylation analyses showed high sensitivity and specificity, which are equivalent or higher than cytology-based screening. However, a uniform assay and population-wide screening are required before drawing further conclusions. DNA methylation–based biomarkers combined with cytology screening might help to improve the sensitivity and specificity of current screening methodology in cervical cancer. Further studies are required to translate these DNA methylation markers for early detection and prognosis of cervical neoplasia for better clinical management.
Footnotes
Acknowledgements
The authors thank TIFAC-CORE for pharmacogenomics study and DST-FIST, Government of India, for infrastructure support; Manipal University and CSIR for fellowship under structured PhD program; and the Government of India for senior research fellowship. The authors also thank all the participants for their kind cooperation in this study. Samatha Bhat and Shama Prasada Kabekkodu contributed equally to this work
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.