
Abstract
Epigenetic alterations within human papillomavirus (HPV) and host cellular genomes are known to occur during cervical carcinogenesis. Our objective was to analyse the influence of (1) methylation within two immunostimulatory CpG motifs within HPV16 E6 and E7 genes around the viral late promoter and their correlation, if any, with expression deregulation of host receptor (TLR9) and DNA methyltransferases (DNMT1, DNMT3A and DNMT3B) and (2) global DNA methylation levels within CpGs of the repetitive Alu sequences, on cervical cancer (CaCx) pathogenesis. Significantly higher proportions of CaCx samples portrayed methylation in immunostimulatory CpG motifs, compared to HPV16-positive non-malignant samples, with cases harbouring episomal HPV16 showing decreased methylation compared to those with viral integration. A significant linear trend of TLR9 upregulation was recorded in the order of HPV–negative controls < HPV16-positive non-malignant samples < HPV16-positive CaCx cases. TLR9 upregulation in cases with episomal HPV16 was again higher among those with non-methylated immunostimulatory CpG motifs. Comparison of cases with HPV–negative controls revealed that DNMT3A was significantly downregulated only among integrated cases, DNMT3B was significantly overexpressed among both categories of cases, although at variable levels, while DNMT1 failed to show any deregulated expression among the cases. Global host DNA hypomethylation, also showed a significant linear increasing trend through the progressive CaCx development stages mentioned above and was most prominently higher among cases with episomal HPV16 as opposed to viral integration. Thus, HPV16 and host methylations appear to influence CaCx pathogenesis, with differential molecular signatures among CaCx cases with episomal and integrated HPV16.
Introduction
Oncogenic human papillomavirus (HPV) infection is the major aetiological factor for cervical cancer (CaCx) pathogenesis. 1 Worldwide, as well as in India, HPV16 infection is the most predominant type associated with CaCx2–4 showing higher prevalence over other types. 5 Disruption of E2 gene through integration into the host genome results in loss of E2-mediated repressor activity and activation of the expression of viral oncoproteins E6 and E7. 6 However, subsets of CaCx cases are also recorded to harbour episomal viral genomes with intact E2 gene suggestive of the existence of alternative mechanisms of loss of E2 repressor activity.7,8
The E7 protein (11 kDa) binds with pocket proteins pRb, p107 and p130 and dissociates the E2F family of transcription factors, which subsequently transactivates cellular proteins required for replication (cyclins A and E). E7 also associates with other proteins like histone deacetylases, 9 components of the activator protein 1 (AP1) transcription complex 10 and the cyclin-dependent kinase inhibitors p21 and p27. 11 E7 drives cells through mitosis in differentiating epithelium overcoming the block to cell-cycle progression. 12 The E6 protein (16 kDa) primarily mediates p53 ubiquitination and degradation. 13 This prevents growth arrest or apoptosis in response to E7-mediated cell-cycle entry in the differentiated epithelial layers. The anti-apoptotic role of E6 protein is emphasized further by its association with Bak.14,15 The high-risk HPV E6 protein contains a C-terminal PDZ-binding domain and binds and degrades several cellular proteins containing PDZ domain. 16 This can mediate suprabasal cell proliferation. 17 The high-risk E6 can also activate the catalytic subunit of telomerase (human telomerase reverse transcriptase (hTERT)). 18 Thus, the abilities of high-risk HPV E6 and E7 proteins to associate with the tumour suppressors p53 and retinoblastoma protein (pRB), respectively, serve as a mechanism by which these viral proteins induce cervical carcinogenesis.
Apart from introducing genetic changes, cervical transformation processes induced by HPV infection have also been linked to viral and host epigenetic changes, including DNA methylation and histone modifications. Recent evidence suggests that gene expression levels are modulated by epigenetic mechanisms involving DNA methylation, microRNAs (miRNAs), long non-coding RNAs (lncRNAs) and histone modifications. 19 DNA methylation is an important regulator of gene transcription, and its role in carcinogenesis has been extensively studied in recent times. It is well established that hypermethylation of CpG islands (CGI) within promoters of specific tumour suppressor genes is associated with transcriptional silencing that affects control of cell proliferation. Cancer-related hypomethylation is also a common event, generally occurring in repetitive sequences, retrotransposons and so on as opposed to hypermethylation of CGI within promoters. More than 90% of all 5-methylcytosine lies within the transposons, including Alu, SINE and LINE, which are comparatively rich in CpG dinucleotides and represent more than one-third of human genome. Alu repeats alone constitute 10% of the human genome. 20
It is now evident that viruses can modulate methylation patterns of the host cell genome by interacting with the host epigenetic machinery, thus driving the silencing of host cellular genes. 21 Such viral exposure–based epigenetic variation in human cancer has been observed for hepatitis B virus (HBV) and hepatitis C virus (HCV) infections resulting in promoter hypermethylation of p16INK4a and oestrogen receptors in hepatocellular carcinomas. 22 Epigenetic changes are known to occur in both HPV and cellular genomes during cervical carcinogenesis. It has been proposed that viral DNA methylation could be a mechanism by which HPVs, like many other viruses, are able to camouflage themselves in the host cell, restricting the expression of viral genes and proteins that are indispensable for viral persistence and avoiding the expression of genes associated with immune response. 23 HPV16 contains 112 CpGs, and in fact, some studies have characterized HPV16 methylomes relevant for cervical cancers.7,8,24
Interestingly, unmethylated CpGs are a signature of bacterial and other pathogen-associated DNA and short unmethylated CpG motifs are immunostimulatory. 25 Hence, CpG methylations within such motifs, if any, in the HPV16 genome are likely to succumb to methylation in order to evade immune surveillance. In fact, the occurrence of an immunostimulatory CpG motif within the E6 gene of HPV16 (nt 496–514) has already been reported and TLR9 is able to recognize this. 23 There also exists another such motif within the E7 gene (nt 762–779) of HPV16, proximal to 3′ end of late promoter P670, and taken together, CpG methylations within such regions of the HPV16 genome are likely to be biologically relevant for CaCx pathogenesis. 23
HPVs are dependent on the host methylation machinery for modulating the methylation status of CpGs within the HPV genome. Therefore, it seems relevant to analyse the expression status of the enzymes belonging to the host methylation machinery, that is, the DNA methyl transferases (DNMTs), which are found to be overexpressed in various types of cancers. 26 DNMT1 is specialized to carry out most of the maintenance methylation following DNA replication, whereas DNMT3A and DNMT3B are responsible for de novo methylation during embryogenesis and germ cell development. They are found to be expressed coordinately in most normal tissues. Therefore, profiling the expression of these DNMTs seems relevant for CaCx pathogenesis.
A series of studies conducted in our laboratory over the past few years27,28 point towards the distinctive biological characteristics of cervical cancers29,30 under the impact of episomal HPV16 (the presence of both episomal and integrated, that is, concomitant or purely episomal viral genomes), as opposed to pure integration. An earlier study from our laboratory 27 revealed an over-representation of methylation at nucleotide 58 within E2BSI among CaCx cases harbouring episomal HPV16 as compared to HPV16-positive normal samples. A subsequent study from our laboratory revealed significantly higher methylation at nucleotide 58 or other CpGs in E2BS-I/II among CaCx harbouring episomal (pure or concomitant) viral genomes as opposed to CaCx cases with purely integrated HPV16 genome. 28
In continuation of our previous studies, herein, we aimed to analyse the methylation status of the two viral immunostimulatory motifs of HPV16 (in E6 and E7 genes) and host receptor (TLR9) expression in various categories of cervical samples, representative of CaCx development. Simultaneously, we also checked the expression of the DNMTs (DNMT1, DNMT3A and DNMT3B) and estimated the extent of global DNA methylation of the host within the repetitive Alu sequences as the surrogate marker among such samples in order to decipher the impact of such factors on cervical cancer pathogenesis.
Results
Analysis of CpG methylation within immunostimulatory motifs of E6 and E7 genes around the late promoter P670
We determined the methylation status of the immunostimulatory motifs located within E6 and E7 regions of HPV16 on either side of the late promoter P670, in view of the possible relevance of such motifs in immune evasion of the host by the virus. A total of seven CpG sites were interrogated by employing bisulphite-modified DNA samples followed by Sanger sequencing, covering the region depicted in Figure 1. All samples showing methylation within the immunostimulatory motifs, that is, at least within CpG sites at nucleotide positions (nt) 502 and 506 for E6, nt 765 and the proximal 780 for E7, with or without methylation at the other CpGs, were scored as methylation positive. The proportion of CaCx cases harbouring methylation in the late promoter was significantly higher for both E6 and E7 immunostimulatory motifs (38/80, 47.50%; p = 0.020 and 70/87, 80.45%; p = 0.036, respectively) compared to HPV16-positive non-malignant samples (5/24, 20.83% and 8/19, 42.10%, respectively). Thus, CpG methylations around the late promoter appeared to be significantly overrepresented among the CaCx cases as opposed to the HPV16-positive non-malignant samples. Representative electropherograms are depicted in Supplementary Figure 1.

Upper panel: bar diagram comparing percentage of samples methylated at different CpGs within the late promoter of HPV16 genome. Lower panel: the immunostimulatory CpG motifs within E6 and E7 regions on either side of the late promoter P670 and a diagrammatic representation of the 7 CpGs analysed at positions 494, 502, 506, 752, 757, 765 and 780.
Among the case samples, for those with data available on 7 CpGs (494, 502, 506, 752, 757, 765 and 780) inclusive of the E6 and E7 motifs, further analysis was undertaken on the basis of their viral physical status (episomal or integrated), and such data are presented in Table 1. A total of 49 CaCx cases with episomal HPV16, 17 CaCx cases with integrated HPV16 and 18 HPV16-positive non-malignant samples were available for such analysis. Overall, proportion of integrated cases with methylation at such CpGs was higher, compared to those with episomal HPV16, as shown in Figure 1. Also, a significant linear trend of progressively higher proportion of methylated samples through HPV16-positive non-malignant samples, CaCx cases with episomal and integrated HPV16 genomes, was recorded for CpG positions such as 494, 502, 506 and 780 as shown in Supplementary Figure 2. The difference between the two CaCx types with respect to methylation at the CpGs around the late promoter was more prominent in case of the three CpGs (494, 502 and 506) towards the 5′ end of P670. This was despite the fact that at all such positions, including CpG position 780, we recorded significant trend of progressively increasing proportion of methylated samples through HPV16-positive non-malignant samples, CaCx cases with episomal and integrated HPV16 genomes. Thus, a significant trend of decreased proportion of samples showing methylation at all such positions was evident among the cases with episomal HPV16 as opposed to those with integrated HPV16.
Percentage of methylation in 7 CpGs along the late promoter region among various categories of cervical samples.

TLR9 expression, HPV16 physical status and immunostimulatory motifs in E6 and E7 genes
Our next attempt was to check whether methylation within the immunostimulatory motifs of E6 and E7 regions of HPV16 was associated with the expression of the innate immune pathway gene TLR9, and if so, whether TLR9 expression was dependent on the viral physical status. We recorded TLR9 expression among 40 out of 103 case samples (38.83% samples) and 15 out of 31 HPV16-positive control samples (48.38% samples) and 7 out of 44 HPV-negative control samples (15.90% samples). Thus, TLR9 expression was significantly overrepresented (p = 0.002) among the HPV16-positive samples. Furthermore, considering samples that expressed TLR9, mean ΔCt values of TLR9 expression for HPV-negative controls (n = 7), HPV16-positive non-malignant samples (n = 15) and HPV16-positive cases (n = 40) were 13.0880, 11.4852 and 11.4487, respectively, reflecting a quantitative increase in such expression among the HPV16-positive samples. TLR9 expression was significantly higher among case samples (p = 0.006; t-test) compared to HPV-negative controls (2−ΔΔCt = 3.1951). TLR9 expression was also significantly (p = 0.018; t-test) higher among the HPV16-positive non-malignant samples (2−ΔΔCt = 3.0373) than the controls. Linear regression analysis showed a significant increasing trend of TLR9 expression (p value < 0.05) from HPV-negative controls to HPV16-positive non-malignant samples followed by HPV16-positive CaCx cases (irrespective of viral physical status) as depicted in Figure 2(a).
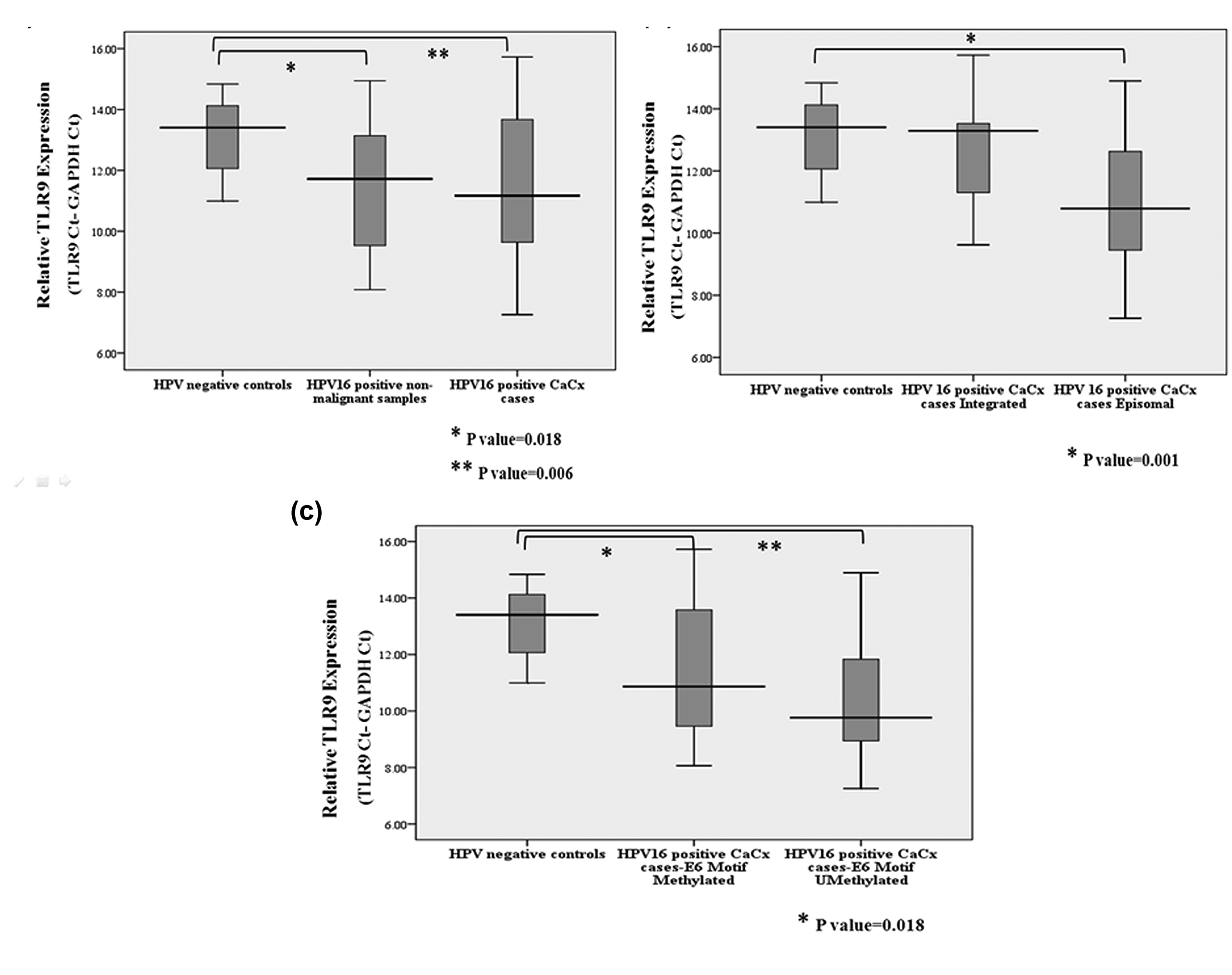
TLR9 expression among different categories of cervical samples normalized to GAPDH expression (lower ΔCt means higher expression). (a) Box plot showing TLR9 expression among HPV-negative controls, HPV16-positive non-malignant samples and HPV16-positive CaCx cases (irrespective of viral physical status). (b) TLR9 expression among HPV-negative controls and HPV16-positive CaCx cases with episomal and integrated viral genomes, and (c) TLR9 expression among HPV-negative controls and cases with HPV16 E6 immunostimulatory CpG motif methylated or unmethylated (irrespective of viral physical status).
Among the 40 case samples analysed for TLR9 expression, viral physical status was available for 35 samples, of which 26 harboured episomal HPV16 and 9 had integrated viral form. Mean ΔCt values of TLR9 expression for the episomal and integrated cases were 11.0050 and 12.5882, respectively. Compared to HPV-negative controls and based on 2−ΔΔCt values, TLR9 expression was 4.2368-fold higher among HPV16 episomal cases and this was statistically significant (p = 0.001). We failed to record any significant variation in TLR9 expression among the integrated CaCx cases compared to HPV-negative controls, as shown in Figure 2(b).
Data on E6 motif methylation were available for a subset of 25 CaCx cases for which, TLR9 expression status was available. The E6 motif was found to be methylated in 48% (12 out of 25) of such cases, while the remaining 52% (13 out of 25) were unmethylated in the E6 motif. Mean ΔCt values of TLR9 expression for these two sets were 11.4102 and 10.4921, respectively. Thus, based on 2−ΔΔCt, TLR9 expression was higher by 3.1993 folds (p = 0.018) among cases with E6 motif methylation, as opposed to 6.0456 folds higher (p = 0.001) among cases without E6 motif methylation in comparison to HPV-negative controls, as shown in Figure 2(c). Among the 25 case samples used for such analysis, HPV16 physical status was available for 23 samples, of which 18 harboured episomal HPV16, while 5 portrayed integrated HPV16. There was a significant trend (ptrend = 0.016) of over-representation of unmethylated E6 motifs (11/18; 61.11%) among the episomal cases as opposed to those of integrated cases (0/5; 0%). Taken together, this was suggestive of the impact of methylation within immunostimulatory motifs of HPV16 on the expression of the innate immune pathway gene TLR9 based on the physical status of the viral genomes.
Expression of host DNMTs, HPV16 physical status and immunostimulatory motifs within E6 and E7 genes
The association of HPV16 early promoter methylation recorded in our previous studies27,28 and that of the late promoter in this study evoked the need to check the expression status of the DNA methylating enzymes DNMTs in CaCx pathogenesis. Mean ΔCt values of DNMT3A expression for HPV-negative controls (n = 19), HPV16-positive non-malignant samples (n = 22) and HPV16-positive cases (n = 36) were 8.0991, 8.1394 and 8.8661, respectively. DNMT3A expression failed to show any significant alteration among the cases or the HPV16-positive non-malignant samples, compared to HPV-negative controls as depicted in Figure 3(a). However, significant downregulation (mean ΔCt = 9.7123, 2−ΔΔCt = 3.0592 and p = 0.001) was prominent among the CaCx cases harbouring integrated HPV16, but no change was obvious among the cases with episomal viral genomes.
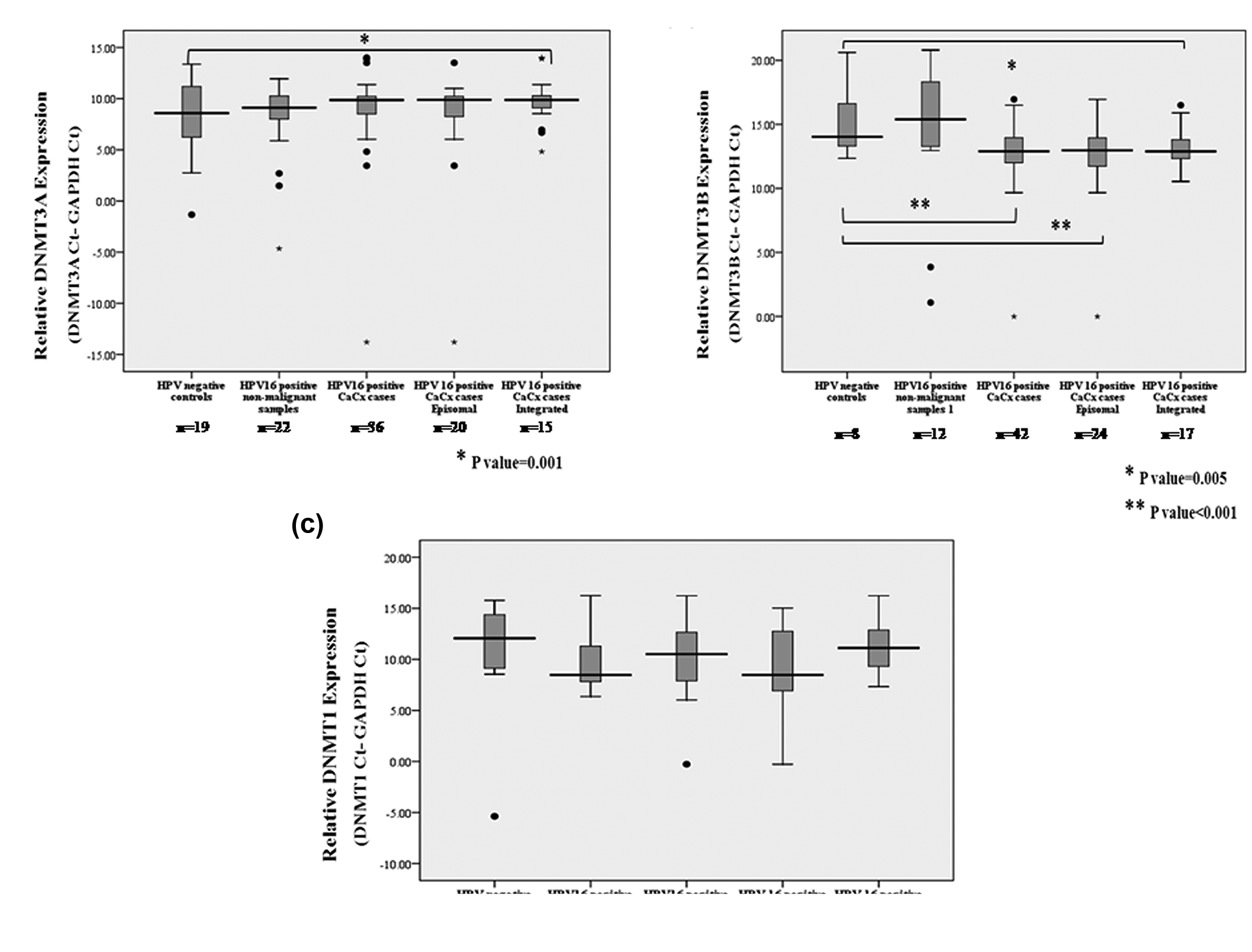
Box plot showing expression of (a) DNMT3A, (b) DNMT3B and (c) DNMT1 among different categories of cervical samples normalized to GAPDH expression (lower ΔCt means higher expression).
In case of DNMT3B, mean ΔCt values for HPV-negative controls (n = 8), HPV16-positive non-malignant samples (n = 12) and HPV16-positive cases (n = 42) were 15.1083, 14.2472 and 12.875, respectively. Analysis revealed that such expression was upregulated by 4.7017 fold (p < 0.001; t-test) among the CaCx cases in comparison to HPV-negative controls, as depicted in Figure 3(b). Such upregulation was more prominent among the CaCx cases with episomal HPV16 (mean ΔCt = 12.4845, 2−ΔΔCt = 6.1637 and p < 0.001) in comparison with those harbouring integrated HPV16 (mean ΔCt = 13.3011, 2−ΔΔCt = 3.4996 and p = 0.005).
DNMT1, on the other hand, failed to show any significant change in expression among CaCx cases (n = 37) or cases with episomal (n = 20) or integrated (n = 16) HPV16, in comparison to HPV-negative controls (n = 8; mean ΔCt = 10.1969) or HPV16-positive non-malignant samples (n = 12; mean ΔCt = 9.7143) as shown in Figure 3(c). Interestingly, HPV16-positive non-malignant samples failed to portray significant changes in the expression of all the DNMTs analysed in comparison to HPV-negative controls.
Having ascertained the deregulated expression of DNMTs in CaCx cases, in association with viral physical status, our next attempt was to check whether methylation within the immunostimulatory motifs of E6 and E7 regions of HPV16 was associated with the expression of the DNMTs. Mean ΔCt values of DNMT3A expression for HPV negative controls (n = 19), case samples with methylated E6 motif (n = 9) and those with unmethylated E6 motif (n = 7) were 8.0991, 6.5431 and 9.9986, respectively. Analysis revealed that such expression was upregulated by 2.9403 fold (p = 0.006; t-test) and downregulated by 3.7308 fold (p = 0.003; t-test) among the CaCx cases with methylated E6 motif and unmethylated E6 motif, respectively, in comparison to HPV-negative controls.
In case of DNMT3B, mean ΔCt values for HPV negative controls (n = 8), case samples with methylated E6 motif (n = 11) and with unmethylated E6 motif (n = 3) were 15.1083, 11.5613 and 13.9256, respectively. Analysis revealed that such expression was upregulated by 11.688 fold (p < 0.000; t-test) among the CaCx cases with methylated E6 motif in comparison to HPV-negative controls. DNMT3B expression failed to show any significant alteration among the cases with unmethylated E6 motif as compared to HPV-negative controls.
Similar analysis failed to reveal any alterations in DNMT1 expression among the cases with respect to the E6 immunostimulatory motif methylation status.
Global methylation status of host genome and HPV16 physical status
The association of expression deregulation of DNMT3B with CaCx cases, irrespective of the viral physical status, and DNMT3A specifically among the cases harbouring integrated HPV16, prompted us to undertake analysis of the global DNA methylation status of the host genome, in relation to CaCx pathogenesis, taking into consideration the physical status of HPV16 genomes. Median percent methylation rate (PMR) values for the various categories of cervical samples such as HPV-negative controls (n = 44), HPV16-positive non-malignant samples (n = 43) and HPV16-positive cases (n = 104) were 44.83%, 41.20% and 13.28%, respectively. A significant trend of increasing hypomethylation was recorded through HPV-negative controls, HPV16-positive non-malignant samples and HPV16-positive CaCx cases (p < 0.001). The median PMR values corresponding to global methylation was significantly lower among HPV16-positive CaCx cases, compared to HPV-negative controls (p < 0.001), as shown in Figure 4(a). Among the 104 case samples, 74 samples characteristically harboured episomal (episomal and concomitant, n = 59) and purely integrated (n = 15) HPV16 genomes, with median PMR values of 9.80% and 22.62%, respectively. In comparison with HPV-negative controls, the median PMR values corresponding to CaCx cases with episomal HPV16 (PMR = 9.80%) and integrated HPV16 (PMR = 22.62%) were both significantly lower (p < 0.001 and 0.004, respectively) as shown in Figure 4(b). Thus, decreased global methylation was most prominent among the cases with episomal HPV16 in comparison to those with integrated HPV16 and such difference was also statistically significant (p = 0.046).

Box plot showing host global DNA CpG methylation status among different categories of cervical samples, based on quantification of CpG methylation within Alu repeats and normalized to β actin CpG methylation expressed in terms of PMR (percent methylation rate) value.
On subsequent analysis, we failed to identify any correlation between such global DNA hypomethylation and expression status of TLR9 and/or DNMTs either based on consideration of the viral physical status or irrespective of such status. Thus, global DNA hypomethylation at repeat sequences of the host might not be directly associated with alterations in expression of TLR9 or DNMTs in such cancers.
Discussion
We undertook this study as a part of our broader objective to test the hypothesis that epigenetic changes (CpG methylation) in HPV16 and host genomes are crucial for the development of HPV16-related CaCx and jointly influence disease risk by modulating the expression of key viral genes and host genes of relevant cellular pathways. Our study provided some insights on the nature of host pathogen interactions with respect to viral and host epigenetic alterations.
Overall decreased methylation within the late promoter region of HPV16 in CaCx cases, specifically those with episomal HPV16, could possibly be biologically insightful. Of such methylations, one at CpG 502 was significantly underrepresented only among cases with episomal HPV16 and this was found to encompass human transcription factor TAFII250 binding site using TRANSFAC database. This happens to be the largest component of the basal transcription factor IID (TFIID), and its binding to the promoter region initiates the assembly of a functional transcription initiation complex. 31 In fact, an earlier study from our laboratory 32 identified that CaCx cases with episomal HPV16, but not integrated HPV16, portrayed expression of L2. Thus, such decreased methylation within the late promoter, concomitant with expression of L2 in such episomal CaCx cases as recorded in our previous study, 32 intrigued us further to predict that such phenomenon could be one of the various mechanisms employed by episomal HPV16 towards expression of the viral late gene L2.
An overrepresentation of viral DNA methylation within immunostimulatory CpG motifs in E6 and E7 regions of CaCx cases, as opposed to HPV16-positive non-malignant samples, was suggestive of the relevance of immune evasion by HPV16 in CaCx pathogenesis employing such CpG motifs. Moreover, higher methylation of such motifs within CaCx cases portraying integrated viral genomes, as opposed to those with episomal HPV16, is further indicative of the fact that such CaCx subtypes are likely to be associated with variable host responses. The host invariably responds to infecting viruses by activating its innate immune system and mounting virus-specific humoral and cellular immune responses, so as to control viral replication and eliminate the infecting virus from the host. 33 Lower methylation within the immunostimulatory motifs of cases with episomal HPV16 implicates that such CaCx subtypes are likely to be less efficient in mounting immune evasion than the ones with integrated HPV16. However, such cases harbour higher viral load concomitant with higher E7 expression, compared to cases with integrated HPV16 as established in an earlier study from our laboratory. 34 This is suggestive of the fact that the episomal cases are unlikely to be less efficient in immune evasion than the integrated cases, thereby evoking the need for further exploration of immune response strategies adopted by the two CaCx subtypes. We, therefore, made an attempt to analyse the expression levels of the endosomal Toll-like receptor, TLR9, which is known to recognize CpG-rich viral DNA and initiate signalling pathways that induce immune response. 35
Concurrent to increased methylation in the immunostimulatory motifs of HPV16 in CaCx cases, we also recorded a significant trend of increasing TLR9 expression among the cervical samples in the following order: HPV-negative control < HPV16-positive normal < HPV16-positive CaCx cases among such histopathologically confirmed cervical tissue samples. This was indicative of the fact that stimulation of TLR9 expression could play a role in CaCx development and could serve as an early risk marker of cervical carcinogenesis, similar to that proposed in an earlier study 36 on cervical cancers and a recent report 37 on pancreatic transformation. Also, TLR9 overexpression among the cases was in line with some recent reports on cervical cancers 37 including a study that employed CaCx cases from an area in the vicinity of our study 38 and other cancers. 39 However, our findings were contradictory to a report, 40 which depicted loss of TLR9 expression among cervical tumour samples as compared to normal cervical samples. Such difference could possibly be attributable to small sample size of eight each in the two groups employed by Hasan et al. 40 in contrast to a large sample set of 103 CaCx cases, 31 HPV16-positive non-malignant samples and 44 HPV-negative controls employed in our study, and/or to ethnic diversity of the two populations.
Interestingly, the fold change of TLR9 over expression was greater among cases harbouring non-methylated immunostimulatory motifs, most prominently among cases harbouring episomal viral genomes and characterized by higher viral copy numbers, as opposed to those with viral integration. 34 This appears to be the first such report on cervical cancers to highlight the differential expression of TLR9 in HPV16-positive CaCx subtypes, and higher levels of TLR9 among the episomal CaCx cases could potentially be attributable to viral load and/or viral antigen–induced stress.
TLR9 is also expressed in the interstitial fibroblast-like stromal cells as well as in cancer cells, and this could be attributable to low rate of metastasis 41 in cancers. In case of breast cancers, 42 high tumour TLR9 expression has been identified to predict good prognosis among triple-negative breast cancer (TNBC) patients. On the contrary, high expression of TLR9 in a variety of solid tumours, such as gastric, colon, ovarian, prostate 39 and cervical cancers, 37 appears to be suggestive of the oncogenic or tumour promoting role of TLR9. In fact, Li et al. 43 showed that in addition to their function of activating immune response, TLRs establish a suitable microenvironment for tumour cell growth, which allows tumour cells to evade immune cells, infiltrate and metastasize, and undergo malignant progression. Thus, overexpression of TLR9 recorded in cervical cancers, 37 which includes our observation as well, appears to be suggestive of a tumour-promoting role of TLR9. Therefore, differential expression of TLR9 among the CaCx subtypes of episomal and integrated HPV16 could potentially be indicative of differential oncogenic status of such cancers.
The host encoded DNMTs that mediate the CpG methylations in the viral and host DNA are observed to be overexpressed, especially DMNT1 and DNMT3B, in a number of solid and haematological cancers. 44 Increased expression of DNMT1 has also been identified through gene expression profiling in short-term primary cervical cancer cultures, in comparison with normal cervical keratinocyte cultures. Genome-wide microarray-based comparative genomic hybridization analysis has also revealed an increase in DNMT3B copy number in association with changes in messenger RNA (mRNA) expression in all HPV-immortalized cell lines and in most cervical cancers analysed. 45 HPV16 E7 is known to associate in vitro and in vivo with the DNMT1. 46 A recent study by Lechner et al. 47 showed increased mRNA expression of both DNMT3A and DNMT1 in HPV-positive head and neck cancer cell lines. Such observations prompted us to explore the expression status of the three DNMTs in HPV16-positive CaCx cases, as compared to HPV-negative controls.
We recorded that DNMT1 expression levels neither differed between cases and controls nor between the two CaCx subtypes. However, significant overexpression of DNMT3B was evident among the cases, which seemed to be higher among the episomal cases as compared to the integrated cases. Studies by Leonard et al. 48 and Au Yeung et al. 49 have revealed that HPV16 can upregulate the expression of DNMT1 and DNMT3B. Thus, upregulation of DNMT3B among the episomal CaCx cases could probably be attributable to higher levels of E7 among such cases. 28 However, lack of alteration in the expression of DNMT1 among the CaCx cases calls for further studies to draw meaningful insights attributable to such observations.
DNMT3A expression, on the other hand, did not differ between the cases and HPV-negative controls, but the integrated subtype portrayed a significantly decreased DNMT3A expression upon such analysis. Although loss of the de novo DNA methyltransferases DNMT3A and DNMT3B in embryonic stem cells is known to obstruct differentiation, the role of these enzymes in somatic stem cells is largely unknown. 50 Mutation in DNMT3A or downregulation of DNMT3A mRNA has also been recorded in a variety of solid tumours, 51 highlighting DNMT3A as a tumour suppressor. Such reports thereby have a significant bearing on the role of DNMT3A as a tumour suppressor in case of CaCx cases with integrated HPV16, as recorded by us. A recent study by Jia et al. 52 identified that ubiquitin PHD RING finger (UHRF) family proteins are frequently upregulated in cancers, leading to downregulation of DNMT3A protein that results in DNA hypomethylation, specifically within highly repetitive sequences.
In fact, re-analysis of the expression data of DNMTs revealed that both DNMT3B and DNMT3A were upregulated among the cases portraying E6 motif methylation, and DNMT3A was downregulated among cases with unmethylated or hypomethylated E6 motifs, in comparison to HPV-negative controls. Such observations clearly reinforce the significant role played by some of the DNMTs in regulating viral DNA methylation as well. Worth noting is the fact that there was an over-representation of CaCx cases harbouring episomal HPV16 genomes among the cases that revealed E6 motif hypomethylation and an over-representation of cases with integrated HPV16 among cases that portrayed E6 motif methylation. Thus, our study appears to be the first of its kind to depict an association of viral DNA methylations with the alteration of the expression status of DNMTs as well, dependent on the viral physical status.
It is now well established that cancer-linked epigenetic changes in DNA involves both cancer-associated DNA hypomethylation and hypermethylation across the genomes. 53 In case of cervical cancers, Kim et al. 54 were the first to report that global DNA hypomethylation increases progressively in high-grade cervical dysplasia and carcinoma in comparison to low-grade dysplasia or normal cervical tissues. Thus, global DNA methylation may serve as a biochemical marker of cervical neoplasia. A number of studies have also identified global hypomethylation as an early event in various cancers such as colon, breast cancer as well as chronic lymphocytic leukaemia (CLL). 55 A direct link between global DNA hypomethylation and genomic instability56,57 is also well established. Taken together, such findings impressed upon us to determine the association, if any, of global DNA hypomethylation with cervical cancer pathogenesis. Our study revealed a significant linear trend of progressive DNA hypomethylation through the discrete stages of CaCx development in the following order: HPV-negative controls > HPV16-positive non-malignant samples > HPV16-positive CaCx cases. This observation was thereby indicative of the fact that DNA hypomethylation could serve as an early marker for singling out HPV16-positive women at risk of developing CaCx in the long run, for which further prospective follow-up studies are warranted.
Global DNA hypomethylation observed so frequently in cancers has been attributed to hypomethylation within highly repeated DNA sequences, which comprise almost half of the human genome.56,57 Alu repeats are the most abundant family of repeats in the human genome, with over 1 million copies comprising 10% of the genome. 20 Alu repeat sequences are often employed to estimate global DNA methylation, 58 and we employed a TaqMan-based quantitative assay to determine the status of DNA hypomethylation in various categories of cervical samples. Thus, hypomethylation appeared to be significantly higher among the CaCx cases, more prominently among the CaCx cases harbouring episomal HPV16, as opposed to those with viral integration. Therefore, such observations prompt us to propose that CaCx cases with episomal viral genomes are likely to portray higher genomic instability than those harbouring viral integration under the impact of higher levels of E7, with respect to Alu repeats’ hypomethylation status. However, such findings need to be confirmed through high throughput assays of whole genome methylation profiling of the host. In fact, genomic instability associated with CaCx cases harbouring integrated viral genomes have been shown by recent study 59 employing high throughput genome sequencing. The study 59 identified HPV integrants flanking and bridging extensive host genomic amplifications and rearrangements, including deletions, inversions and chromosomal translocations. Therefore, it seems likely that patterns of genomic instability could be divergent among the two subtypes of CaCx cases. Thus, hypomethylation of CpGs within Alu repeats could be one of the mechanisms adopted by CaCx cases with episomal HPV16 to maintain the cancer phenotype of genomic instability, besides other mechanisms.
DNA methyltransferases are key players in establishment and maintenance of epigenetic control. DNMTs function through the interaction with a host of other proteins, including those of the chromatin components, transcription factors, replication and repair machinery. Therefore, it becomes difficult to predict the impact of DNMTs on DNA methylation through mRNA expression level studies. However, as discussed above, downregulated levels of DNMT3A protein under the impact of UHRF family proteins have been attributable to DNA hypomethylation 52 in cancers. In fact, our study clearly demonstrated that decreased DNMT3A expression is associated with HPV16 E6 motif hypomethylation, which is characteristically recorded among episomal CaCx cases portraying higher DNA hypomethylation at the Alu repeat sequences as well. This is in contrast to cases with integrated HPV16 portraying higher E6 motif methylation, lower hypomethylation within Alu repeat sequences and upregulation of DNMT3A. Therefore, our observation of significant downregulation of DNMT3A at the mRNA level only among the CaCx cases harbouring integrated HPV16, irrespective of the consideration of the viral E6 motif methylation status, probably does not reflect the actual scenario. However, failure to detect any direct correlation between DNMTs and global DNA hypomethylation within the repeat sequences among such HPV16-positive CaCx cases could be attributable to a number of factors. Some such potential factors associated could be activation of demethylating enzymes, formation of repressive chromatin domains and progressive loss of DNA methylation due to faster growth of cancer cells, slowing down the copying of methylation from replicating parental DNA as proposed in case of breast cancers. 60
In conclusion, our study highlighted the significant role of DNA methylations at biologically relevant regions of both HPV16 and the host in cervical cancer pathogenesis with a clear demonstration of host pathogen interactions. We have shown that HPV16-positive cervical cancer samples showed decreased global methylation compared to the HPV-negative controls, which was more prominent in episomal cases that also revealed E6 motif hypomethylation as compared to the integrated CaCx cases. Clearly, in line with our previous studies,27,28,34,61 we further establish that CaCx cases with episomal and integrated viral genomes are biologically distinct entities and are likely to portray differential molecular signatures that could perhaps differentiate between the two subtypes at the level of disease prognosis as well.
Materials and methods
Samples and subjects
The samples used for this study were incorporated from another ongoing natural cohort study.27,61–64 The malignant samples selected for the study were histopathologically confirmed invasive squamous cell carcinomas of tumour stage III and above and majority were diagnosed as moderately differentiated squamous cell carcinomas. These were derived from married subjects (median age: 52 years; range = 23–80 years) attending a cancer referral hospital (Saroj Gupta Centre and Research Institute, South 24 Parganas, West Bengal, India) during the period of 1998–2013.
The non-malignant samples were normal cervical scrapes confirmed by Pap smear test and derived from married and non-pregnant (or 6 months post-partum) women (median age: 33 years; range = 24–58 years) with no previous history of cervical dysplasia/malignancy. All such samples were collected during the period of 1998–2010. We also collected non-malignant biopsy samples from disease-free married individuals undergoing hysterectomy for various reasons other than cancers such as prolapsed, fibroid and cyst for our study.
All samples, cases and non-malignant group samples were collected from the subjects with informed consent approved by the institutional ethical committee for human experimentation. Details regarding subjects, samples, DNA isolation, HPV screening and determination of HPV16 genome physical status, RNA isolation and c-DNA preparation are described earlier from our laboratory.27,28,32,61–64
Determination of methylation status of the CpG sites in the late promoter region, containing two immunostimulatory motifs
For such assay, the modified DNA was amplified using primers BSHPV16-2F and BSHPV16-2R (positions 419–691) and BSHPV16-3F and BSHPV16-3R (positions 668–971). 24 First, a polymerase chain reaction (PCR) was done with BSHPV16-2F and BSHPV16-3R primers and then two nested PCRs were done with BSHPV16-2F/BSHPV16-2R and BSHPV16-3F/BSHPV16-3R with the product of first PCR. For the first PCR, the reaction mixture (25 µL) contained 10 ng modified DNA, 70 ng primers, 50 mM KCl, 2 mM MgCl2, 50 mM Tris-HCl (pH 8.3), 50 mM (NH4)2SO4, 150 µM of each deoxynucleotide triphosphate (dNTP) and 2 U of FastStart DNA Polymerase (Roche Diagnostics). For the nested PCR with BSHPV16-2F/BSHPV16-2R, the reaction mixture (25 µL) contained 1 µL long PCR product, 50 ng primers, 50 mM KCl, 2 mM MgCl2, 50 mM Tris-HCl (pH 8.3), 50 mM (NH4)2SO4, 150 µM of each dNTP and 1.25 U of FastStart DNA Polymerase. For the nested PCR with BSHPV16-3F/BSHPV16-3R, the reaction mixture (25 µL) contained 1 µL long PCR product, 60 ng primers, 50 mM KCl, 2 mM MgCl2, 50 mM Tris-HCl (pH 8.3), 50 mM (NH4)2SO4, 60 µM of each dNTP and 1.25 U of FastStart DNA Polymerase. The primer sequences, amplicon size and PCR conditions are provided in Supplementary Table. PCR products were verified by running on agarose gel and direct sequencing was carried out using primers BSHPV16-2F and BSHPV16-3F. Sanger sequencing was done in an ABI Prism™ 3100 automated sequencer using Big dye terminator chemistry.
Assessment of TLR9 mRNA expression by quantitative real-time PCR (SYBR Green assay)
TLR9 expression was estimated along with the housekeeping gene GAPDH (Glyceraldehyde-3-phosphate dehydrogenase) as an internal control. The reaction mixture (10 µL), for each reaction, contained 2× Power SYBR® Green PCR Master Mix (Applied Biosystems; SYBR® Green 1 Dye, AmpliTaq Gold® DNA Polymerase LD, dNTPs with deoxyuridine triphosphate (dUTP)/deoxythymidine triphosphate (dTTP) blend, Passive Reference 1 and optimized buffer components), 25 ng primers and complementary DNA (cDNA; synthesized with random hexamer primers). The primer sequences, amplicon size and PCR conditions are provided in Supplementary Table.
Comparative CT method of analysis was followed to find out the fold change in mRNA expression between HPV16-positive cases and HPV-negative controls and HPV16-positive non-tumours. Each assay was replicated at least twice. Three to four non-template controls were included in each assay. Dissociation curve analysis (Supplementary Figure 3) for target gene was performed to verify specific primer-binding. Melting temperatures for PCR products of GAPDH (shown in Supplementary Figure 7) and TLR9 genes were 79.7°C and 86°C, respectively. Specific amplification plot is shown in Supplementary Figure 3 for TLR9 and Supplementary Figure 7 for GAPDH.
Assessment of DNMTs (3A, 3B and 1F) mRNA expression by quantitative real-time PCR (SYBR Green assay)
Expression of DNMT3A, DNMT3B and DNMT1 were determined along with GAPDH as internal control. The primer sequences, amplicon size and PCR conditions are provided in Supplementary Table.
Comparative CT method of analysis was used as in case TLR9 expression study. Dissociation curve analysis (Supplementary Figures 4–6) for the target genes DNMT3A, DNMT3B and DNMT1 were performed to verify specific primer-binding. Melting temperatures for PCR products of DNMT3A, DNMT3B andDNMT1 genes, respectively, were 88°C, 87°C and 81°C. Specific amplification plot is shown in Supplementary Figures 4–6 for DNMT3A, DNMT3B and DNMT1, respectively.
Determination of global methylation status of host
In all, 44 HPV16-negative cytologically normal controls, 43 HPV16-positive cytologically normal controls and 104 CaCx samples were analysed by TaqMan-based quantitative real-time PCR of bisulphite-modified DNA. On modified DNA, TaqMan assay was done. A volume of 5 ng of modified DNA was targeted for quantitative real-time PCR (qRT-PCR) in a reaction mixture of 5 µL containing 25 ng of primers and 0.1 µM of hydrolysis probe 58 in TaqMan Universal Master Mix (PE Applied Biosystems; PerkinElmer). Two different mixes were prepared for each set of primers and β actin was used as an internal control. PMR (percent of methylated reference) value was calculated based on the formula PMR = 2−ΔΔCt × 100%, where ΔΔCt = {Ct of ALU-Ct of β actin} case sample − {Ct of ALU-Ct of β actin}control samples. The primer sequences, amplicon size and PCR conditions are provided in Supplementary Table.
Statistical analysis
Kolmogorov–Smirnov test was performed to identify whether the test variables followed normal distribution. Independent two-sample t-test and Mann–Whitney U test were used to identify association with disease phenotype for variables those followed normal distribution and those that did not, respectively. Pearson correlation test was performed to identify any correlation between two test variables. Chi-squared test was performed to test for association of a variable with disease status. Trend test was performed to identify any trend among test variables. All analyses were performed using software package SPSS for windows v16.0.
Footnotes
Acknowledgements
The authors thank Saroj Gupta Cancer Centre and Research Institute (SGCCRI, Thakurpukur, South 24 Parganas, West Bengal, India), Child in Need Institute (Pailan, South 24 Parganas,West Bengal, India), Calcutta Medical College Hospital (Kolkata, West Bengal, India) and Jawharlal Nehru Medical College Hospital (Kalyani, Nadia, West Bengal, India) for their support in sample collection; all members of Human Genetics Unit, Indian Statistical Institute, Kolkata, India and National Institute of Biomedical Genomics, Kalyani, India for their technical support during the work and specially thank also University Grant Commission (UGC), India for providing a Fellowship (JRF and SRF) to Ms Shrinka Sen and Council of Scientific and Industrial Research (CSIR) for providing a Fellowship (JRF and SRF) to Dr Paramita Mandal to work on this project.
Compliance with ethical standards
Written informed consent was obtained from all individual participants included in the study. The study was approved by the Institutional Review Board of the National Institute of Biomedical Genomics, Kalyani, West Bengal, India.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported partially by Department of Biotechnology (Grant No. BT/PR2012/MED/29/312/2011), Government of India, and partially by National Institute of Biomedical Genomics (Intramural Funding).