
Abstract
Raf-kinase inhibitor protein has been reported to inhibit both the Raf/mitogen extracellular signal–regulated kinase/extracellular signal–regulated kinase and nuclear factor kappa-light-chain of activated B cells pathways. It has also been reported in cancers that Raf-kinase inhibitor protein behaves as a metastatic suppressor as well as a chemo-immunosensitizing factor to drug/immune-mediated apoptosis. The majority of cancers exhibit low or no levels of Raf-kinase inhibitor protein. Hence, the activities of Raf-kinase inhibitor protein contrast, in part, to those mediated by several cancer stem cell transcription factors for their roles in resistance and metastasis. In this review, the existence of crosstalks in the signaling pathways between Raf-kinase inhibitor protein and several cancer stem cell transcription factors (Oct4, KLF4, Sox2 and Nanog) was assembled. Oct4 is induced by Lin28, and Raf-kinase inhibitor protein inhibits the microRNA binding protein Lin28. The expression of Raf-kinase inhibitor protein inversely correlates with the expression of Oct4. KLF4 does not interact directly with Raf-kinase inhibitor protein, but rather interacts indirectly via Raf-kinase inhibitor protein’s regulation of the Oct4/Sox2/KLF4 complex through the mitogen-activated protein kinase pathway. The mechanism by which Raf-kinase inhibitor protein inhibits Sox2 is via the inhibition of the mitogen-activated protein kinase pathway by Raf-kinase inhibitor protein. Thus, Raf-kinase inhibitor protein’s relationship with Sox2 is via its regulation of Oct4. Inhibition of extracellular signal–regulated kinase by Raf-kinase inhibitor protein results in the upregulation of Nanog. The inhibition of Oct4 by Raf-kinase inhibitor protein results in the failure of the heterodimer formation of Oct4 and Sox2 that is necessary to bind to the Nanog promoter for the transcription of Nanog. The findings revealed that there exists a direct correlation between the expression of Raf-kinase inhibitor protein and the expression of each of the above transcription factors. Based on these analyses, we suggest that the expression level of Raf-kinase inhibitor protein may be involved in the regulation of the cancer stem cell phenotype.
Introduction: Raf-kinase inhibitor protein
A phosphatidylethanolamine-binding protein-1 (PEBP1) was identified, cloned, and found to inhibit the Raf-1/mitogen extracellular signal–regulated kinase/extracellular signal–regulated kinase (Raf/MEK/ERK) pathway by binding directly to Raf-1. The PEBP1 protein was retitled the “Raf-kinase inhibitor protein” (RKIP). 1 RKIP is a regulatory molecule expressed across many organisms and tissue types and is involved in various intracellular signaling networks.2,3 Since its discovery, RKIP as a PEBP1 protein has been shown to bind to the opioid morphine-6-glucuronide4,5 among other small hydrophobic ligands. RKIP is the first endogenous protein that can inhibit the mitogen-activated protein kinase (MAPK) cascade through binding to Raf-1 through a direct protein–protein interaction.1,6
In addition to its inhibition of the Raf-1/MEK/ERK pathway, RKIP has been shown to downregulate the G protein–coupled receptor (GPCR) 7 signaling by binding to the G protein–coupled receptor kinase-2 (GRK2) and the nuclear factor “kappa-light-chain-enhancer” of the activated B cells (NF-κB) pathway through binding to upstream regulatory molecules. 8 In fact, there is some crosstalk found between the MAPK pathway and the GRK2 molecule. The intricate control that RKIP maintains over the expression and the intensity of signaling pathways indicates the importance it carries in many cellular processes including the cell cycle, 9 cell transformation, 1 apoptosis, 10 cardiomyocyte contractility, 7 tumor progression, and metastasis suppression,11,12 particularly in prostate and breast cancers.
Experiments performed in RKIP knockout mice demonstrated the impact on behavioral responses and tumor progression. One study showed that the absence of RKIP protein in mice exaggerated the phenotype of hypoxia-induced pulmonary hypertension as RKIP-modulated ERK appears to play an essential role in MAPK signaling and pulmonary arterial hypertension progression. 13 As RKIP is expressed across most tissues and cell types, knocking out RKIP in mice led to a deficit in olfaction from the expression in the brain. 14 The lack of RKIP expression has also been found to accelerate the potency of tumor progression and metastasis in genetic mouse models of prostate cancer. 15
The human RKIP gene is located on chromosome 12q24.23 and consists of four exons with a single major transcript called RefSeq entry NM_002567 187 amino acid protein. The RKIP complementary DNA (cDNA) follows the sequence shown in Table 1. The transcription start site (TSS) of RKIP is 146 base pair (bp) upstream of the ATG site. The use of cap analysis of gene expression (CAGE) allowed the characterization of TSS, 16 where most of the RKIP TSS are in groups from 21 to 99 bases downstream of the RefSeq TSS. 17 The highest confidence rate of matching the 13 PolII core sequence matrices 18 was found 46 bp upstream of the TSS as a CCAAT-box through the matrix-scan program in the RSAT suite. 19 RKIP follows a pattern of dispersed initiation where multiple TSS are scattered in local regions within 50–100 nucleotides of each other, 20 in which the RKIP core promoter lacks a TATA box but has multiple CpG islands. 21 In place of the TATA box, Sp1 binds to two cis-binding sites discovered in tandem near the TSS. 22 Sp1 may play a role in anchoring the constitutive transcriptional regulator to the core promoter through the interaction of the TFIID complex [composed of several subunits called TATA-binding protein Associated Factors (TBP-associated factors, or TAFs)]. 23
CCDS 9187.1 Homo sapiens Chromosome 12 PEBP1 Sequence Data from NCBI database.

cDNA: complementary DNA; NCBI: National Center for Biotechnology Information; PEBP1: phosphatidylethanolamine-binding protein-1.
Blue color indicates alternating exons, and red, splice junction.
RKIP is a 23-kDa protein with 187 amino acids expressed in different species including human, bovine, and plant. Its protein sequence is shown in Table 1. The tertiary globular structure of RKIP contains a hydrophobic cavity that serves as a ligand-binding pocket and as a PE-binding domain which binds to phosphatidylethanolamine. The ligand-binding domain is also involved in the Raf/MEK binding as shown in Figure 1. The pocket space can bind to various ligands only at low pH but not under physiological conditions. 24 These various ligands include O-phosphatidylethanolamine, O-phosphorylserine, O-phosphoryltyrosine, and O-phosphorylthreonine. There is versatile, flexible binding of other small molecules and proteins like nucleotides,25,26 morphine and morphine-glucuronides, 5 and the chemical compound phosphodiesterase-6 inhibitor (sildenafil). 27 Moreover, the pocket binds to cacodylate, 28 which indicates that the phosphogroup binding is possible since cacodylate mirrors the phosphate groups. The ligand-binding pocket is involved in the inhibition of the ERK pathway and plays a role in Raf-1 binding by interacting with the phosphorylated N-region of Raf-1.26,29

The RKIP protein has specific binding sites for MEK that ranges from the 27th amino acid to the 77th and for Raf-1 that includes from the 77th to the 108th amino acid.
In humans, the RKIP structure consists of four alpha-helices and nine beta-strands. A large central beta-sheet consisting of six strands can be viewed by X-ray crystallography in Figure 2. 28 The tertiary structures are shown in correspondence to the amino acid sequence and secondary structures in Figure 3.

The 3D structure of RKIP consists of alpha-helices and antiparallel beta sheets. The circled region indicates the ligand-binding pocket site. Phosphorylation sites of S153 and P74 are indicated on the structure.

The specific sequence chain of RKIP demonstrates the placement of secondary structures in relation to the primary amino acid sequence.
RKIP transcription and regulation
The normal expression of RKIP is found in all tissues in humans and mammals. 21 The RKIP gene uses dispersed transcription initiations which are usually associated with constitutive genes rather than focused transcription initiations. Dispersed initiation uses multiple start sites located across a range of 50–100 nucleotides. A potential enhancer 13 kb upstream of RKIP was found due to multiple transcription factor (TF) binding sites (TFBS) shown in Figure 4 and an advanced level of H3K4RMe4. The CCAAT-box has the highest probability as the binding site for NF-Y proteins, found 46 bp upstream of TSS. RKIP is found to fit the constitutive promoter of RKIP TSS. 30

The RKIP gene is spaced between four exons and three introns on chromosome 12. Various transcription factor binding sites (TFBS) are shown including AP-1, c-Jun, c-Myc, p300, PPAR- γ1, PPAR-γ2, and YY1. The transcription start site is localized past the majority of the TFBS. Not shown are the CpG islands that can also initiate transcription of RKIP.
The identity of the RKIP promoter was narrowed down by the ENCODE project data that characterized the regulatory regions where chromatin modifications and TFBS are localized. The qualifications of a strong promoter around the RKIP 5′ exon include a large intensity of H3K4Me3 modification and several TFBS assayed by chromatin immunoprecipitation sequencing (ChIP-Seq). There is a possible enhancer sequence 13 kb upstream of RKIP that contains multiple TFBS and a high degree of H3K4Me1 and H3K27Ac modifications. 31 The TF CREB (cyclic adenosine monophosphate response element–binding protein) is known to be responsive to the secondary messenger molecule cyclic adenosine monophosphate (cAMP) 32 which induces binding to the single TFBS upstream of the Sp1 binding sites. 22 If GPCR is stimulated, high levels of cAMP are produced in response. High intracellular cAMP levels cause the activation of the protein kinase A (PKA) which then induces the phosphorylation and activation of CREB. 33 p300/chromatin binding protein (CBP), a histone acetyltransferase that acetylates histones in chromatin, is then co-activated with CREB, in which the core RKIP promoter recruits the histone modifier protein p300 to increase RKIP transcription. 32 Histone acetylation loosens DNA conformation for greater levels of transcription. CREB and p300 are important for RKIP expression in transient in an in vitro reporter assay in cancer cells. 22 Therein, RKIP is highly expressed in the nervous system, adrenals, and thyroid and is not highly expressed in white blood cells and cardiac cells. 34
An additional modulator called Snail1 is found to repress the transcription of RKIP at the RKIP promoter. 35 Snail1 is a known regulator of the epithelial–mesenchymal transition (EMT) that promotes invasiveness, anti-apoptotic properties, and migratory ability. 36 By experimenting with loss-of-function and gain-of-function approaches, Beach et al. 35 concluded that Snail1 inhibited RKIP transcription initiation at the proximal E box of its promoter in prostate cancer cell lines. The direct suppression of RKIP by Snail1 suggests that RKIP may play an essential not only in tumor invasiveness and metastatic ability but also in EMT development.
As reported, RKIP is an allosteric inhibitor of MEK and Raf-1 complexes. 37 However, it cannot bind to both Raf-1 and MEK simultaneously since the binding domains overlap as seen in Figure 5. The mapping of RKIP shows one clear binding domain for MEK and multiple domains for Raf-1. Remarkably, when the segment from the BspEI site to the carboxy terminus is removed, the association with Raf-1 is enhanced. However, when any fragment after the PpuMI site is deleted, Raf-1 binding affinity to RKIP is decreased, suggesting that the complex relationship between Raf-1 and RKIP binding involves a main site located in the BspEI–PpuMI fragment binding to amino acids 77–108 38 as seen in Figure 1 as well.

The 187 amino acid sequence of RKIP is given with the precise MEK and Raf-1 binding sites. The phosphorylation at serine 153 is highlighted by the boxed area.
RKIP-mediated inhibition of the MAPK and NF-κB pathways
Under basal conditions, RKIP is normally bound to Raf-1 and cannot be activated by rat sarcoma (Ras). However, when conditions that stimulate mitogen activation, the Ras/Raf/MEK/ERK cascade is initiated. A growth factor will bind to a receptor and starts the downstream signaling cascade, activating Ras, which in turn binds to Raf-1, and the dimeric complex translocates to the membrane. 39 Raf-1 activation induces the downstream signaling cascade, starting with the phosphorylation and activation of the MEK/ERK, which in turn activates the ERK. The overview of the Raf/MEK/ERK signaling pathway is shown in Figure 6. Cytoplasmic ERK carries out a multitude of essential functions including migration to the nucleus to phosphorylate TFs40,41 and binds to promoter sequences.42,43 ERK can upregulate gene expression that is specific to controlling cellular responses such as cell migration, differentiation, and proliferation, and the intensity of the signal is dependent on the positioning and timing of ERK activity. 44 Since ERK is a crucial regulator of cell fate, it is important to have tight and fine-tuned controls over the complexity of RKIP regulation of ERK.45,46

A general overview of the three different signaling pathways interacting with RKIP. RKIP inhibits the MAPK pathway by binding to Raf-1. It can also inhibit NF-κB signaling by inducing IKK to bind IKB to NF-κB. Reversibly, when PKC phosphorylates RKIP, RKIP can bind to GRK2 to allow the activation of GPCR downstream signaling.
RKIP can inhibit Raf-1 by a variety of tested mechanisms. Since the binding domains for Raf-1 and MEK overlap on RKIP, RKIP can only bind to Raf-1 or MEK one at a time, not simultaneously. However, Raf-1 can bind to RKIP on subdomains I and II, while MEK binds to Raf-1 kinase subdomains VIb–VIII. 38 However, steric hindrance impedes RKIP binding to Raf-1 when bound to MEK due to the protein overlap into subdomain I and the N-region. 47 Thus, instead of inhibition by posttranslational modifications, RKIP physically blocks the binding of Raf-1 to MEK by competing with MEK binding, contributing to steric hindrance, or decreasing the binding affinity.
The N-region may play an important role in the precise control of MEK/ERK activation. It has been reported that Raf-1 can be activated by the phosphorylation of S338/S339 and Y340/341 in the N-region of Raf-1.48,49 Mutational in vitro studies have suggested that the phosphorylation of the Raf-1 N-region acts as a docking signal for RKIP to bind to Raf-1 at the appropriate location 46 and maintains Raf-1 in its phosphorylated state.36,46 By this mechanism, RKIP prolongs the phosphorylation of Raf-1 and controls the precise temporal condition of MEK activation through Raf-1 binding. Furthermore, peptide arrays have been used suggesting that RKIP can bind to not only Raf-1 but also to MEK and Ras, thus interfering with the activation of Raf-1.
In addition, by inhibiting the MAPK pathway, RKIP can also induce anti-metastatic properties as studied in metastatic and invasive breast cancer cells. Normally, the MAPK pathway activates V-Myc Avian Myelocytomatosis Viral Oncogene Homolog (Myc), which in turn promotes LIN28 expression and represses Let-7 processing. However, when RKIP blocks the MAPK pathway, it consequently enhances Let-7 processing and thus suppresses High Mobility Group AT Hook-2 (HMGA2), a chromatin remodeler known to activate invasive- and metastasis-promoting genes like Snail. 12 This signaling cascade is one of the mechanisms in which RKIP functions as a metastatic suppressor.
RKIP is also known to inhibit the ERK pathway, but the mechanistic details are vaguely portrayed. Quiescent cells lack MAPK signaling due to RKIP bound to Raf-1. In mitogen-activated cells, RKIP is disassociated which allows the downstream signaling cascade to activate the MEK/ERK pathway until the stimulus wears off and RKIP binds to Raf-1 again. The dissociation between RKIP and Raf-1 is led by protein kinase C (PKC) phosphorylating S153 on RKIP, 50 which in turn enhances MEK activation. The PE-binding pocket can transform to a multitude of different conformations within milliseconds which leads to the hypothesis that the pocket is important for RKIP substrate protein–protein interaction. 51 After a certain duration, ERK-induced negative feedback loops 39 kick in to recruit RKIP and deactivate the signaling cascade. The phosphorylation of S153 by PKC is known not only to promote the dissociation of RKIP to Raf-1 but also to upregulate the dimerization of RKIP and the binding of RKIP to other regulatory molecules such as GRK2. The NF-κB is a TF that upregulates many genes related to cell proliferation, survival, and immunity.52–54 In its inactive state, NF-κB remains as a cytosolic dimeric complex bound by IκB (inhibitor of kappa light chain enhancer of activated B cells). When activated, the two kinase subunits of inhibitors of κB kinase α and β (IκB kinase α (IKKα) and IKKβ) of the IKK complex phosphorylate IκB, resulting in the ubiquitination and degradation of IκB. Thus, the NF-κB is released and translocated into the nucleus to be activated. 55 Key activators of the NF-κB pathway include the pro-inflammatory interleukin factor 1-beta (IL-1β) and the tumor necrosis factor-α (TNF-α). 56
Among its diverse array of functions, studies suggest that RKIP acts as an inhibitor of the NF-κB pathway by binding to the upstream regulatory activators of the NF-κB dimer. RKIP can co-immunoprecipitate with IKKα and IKKβ, and the NF-κB-inducing kinase (NsIK) and the transforming growth factor-β–activated kinase (TAK1). Yeung et al. 8 also performed an NF-κB luciferase reporter assay with a transfected RKIP expression vector and the results showed lower NF-κB activity in the presence of IL-1β and TNF-α. The results suggest the same inhibitory response of RKIP to the NF-κB signaling. The overview of the interaction between RKIP and the NF-κB pathway is shown in Figure 6.
Changes in GPCR signaling
RKIP plays a significant role in regulating molecules involved in the GPCR signaling, specifically the GRK2. GRKs regulate the activation and inhibition of GPCRs as serine and threonine kinases. Their substrates, GPCRs, define the majority of drug-targeted molecules on cell surfaces to deregulate extracellular signaling into the cell. 57 Among the seven kinases that constitute the GRK family, GRK2 falls under the β-adrenergic receptor kinase subfamily. 58 GRK2 is an inhibitor of GPCRs by direct binding and phosphorylation.59,60
Studies in GRK2-depleted knockout mice have suggested GRK2 to play a degenerative role in cardiovascular diseases. The kinase normally responds to catecholamine-induced reactions and controls the myocardium’s contraction and response. In the absence of functional GRK2, β-adrenergic receptors do not respond to interfering stimuli and, thus, are protected in heart failure. GRK2 is being studied further to elucidate its regulation mechanism to gain better insight on its effect on cardiovascular diseases.61–65
With respect to signaling pathways, RKIP can directly interact with GRK2 to upregulate G protein signaling. When RKIP is phosphorylated at S153 by PKC, RKIP dissociates from Raf-1 and binds to GRK2 which consequently enables GPCR signaling. 7 The amino-terminal sequence of GRK2 acts as the binding domain for GRK2 and RKIP 7 which may explain the specificity of RKIP for GRK2 among the other GRK subfamilies. The interaction site of GRK includes an RH domain (regulator of G protein signaling homology domain) and an amino-terminal domain only found in the GRK family. RKIP can specifically target the receptor substrates of GRK2 due to the difference in the N-terminal domain of GRK2. Its unique structural and functional differences can be explained in the GRK2’s RH domain that can bind and gather active Gαq subunits, while other GRKs lack the ability to do so.66–68 The overview of the interaction between RKIP and the GPCR signaling pathway is summarized in Figure 6.
RKIP expression in cancer tissues
Decreased RKIP expression levels generally have been associated with multiple cancer types and tumorigenesis. Loss of RKIP typically accelerates the cancer progression, while overexpression of RKIP normally impedes tumorigenesis and metastasis. While a few studies have distinguished the effects of RKIP and phosphorylated Raf-kinase inhibitor protein (pRKIP), most reported studies have not investigated the difference between the active and inactive forms of RKIP.
In B-cell lymphoma, RKIP works to decrease the response of activated Raf, and increased RKIP expression has been associated with increased anti–CD20 therapy-induced B-cell apoptosis. 69 In addition, high levels of RKIP have a strong correlation to the sensitization of Ramos B-cells to the tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) caused by a chimeric anti–CD20 mouse/human monoclonal antibody. 70 RKIP has been associated with the sensitization of B-cell lymphoma tissue to apoptosis.
In bladder cancer, the transcriptional regulation of RKIP mRNA is decreased in transitional cell carcinoma (TCC) compared to normal cells. 71 Translational regulation of RKIP expression determined the degree of cancer progression and lymphatic vessel invasion. Additional studies with bladder cancer have shown that decreased RKIP levels may correlate to an invasive phenotype. 72
In brain cancer, the downregulation of RKIP expression is found mainly in gliomas and glioblastomas.73–75 RKIP may play a role as a biomarker in brain cancers as it often marks malignant progression and decreased expression of RKIP is associated with cell migration and metastasis. 75
In breast cancer, downregulation of RKIP is seen in invasive carcinoma and complete loss of RKIP is apparent in lymph node metastases. 76 Studies have shown consistent results of RKIP being a metastasis suppressor77,78 where decreased RKIP expression promotes metastasis and increased RKIP expression inhibits cancer metastasis and invasiveness in the MDA-MB-231 breast cancer cell line. 79 Another finding shows RKIP as a regulator of stromal genes to impede breast cancer bone metastasis. 80 Upregulation of RKIP also induces apoptosis of cancer cells undergoing chemotherapy. 10
In colorectal cancer (CRC), RKIP expression is downregulated in CRC tissue compared to normal mucosa and is decreased even further in metastatic CRC cells. 81 Reduction of RKIP is also found in association with metastasis and vascular invasion. 82 Regulation of RKIP is affected by the CpG island methylator phenotype (CIMP) 83 that contributes to the reduced levels of RKIP along with other tightly controlled mechanisms in CRC.
In esophageal cancer, decreased RKIP expression is found in the majority of esophageal squamous cell carcinoma patients84–86 and is associated with poor prognosis. 87 The discovery of the RKIP promoter being hypermethylated suggests the transcriptional activity of RKIP at the genetic level contributes to diminished RKIP expression. 88
RKIP expression in gastric cancer is found to be downregulated compared to the expression in normal tissues.89–92 Further downregulation of RKIP is found in lymph node metastases of gastric cancer patients 93 which confirms the role of RKIP as a metastasis suppressor protein. Hypermethylation of RKIP may be conducive to particular branches of gastric cancer.89,94 Interestingly, in gastric adenocarcinoma (AGS) cells, Helicobacter pylori can induce phosphorylation of RKIP at S153 and also mutate the serine to valine to disable RKIP’s structure and Raf-1-related function. 95 In addition, H. pylori–induced apoptosis requires H. pylori–mediated phosphorylation of RKIP possibly through the mammalian target of rapamycin (mTOR) pathway. 95 H. pylori infection is instrumental in threatening RKIP expression in AGS.
Ovarian cancer (OVCA) cells have greatly diminished RKIP levels compared to normal tissue expression and have increased invasive potentiality which suggest that RKIP to be a metastasis suppressor gene. 96 Upregulation of RKIP expression inhibits OVCA metastasis and progression. 97 Decreased RKIP expression is also found in cervical cancer98,99 and is associated with increased metastatic potential, greater ability of tumor vascularization, and apoptosis resistance in cisplatin chemotherapy. 99 Curiously, RKIP expression is diminished in endometrial cancer but has no observable clinical importance. 91
RKIP expression is downregulated in hepatic cell carcinoma (HCC), 100 most likely by methylation. 101 Upregulation of RKIP inhibits both HCC tumor growth and cell migration. 100
In lung cancer, decreased RKIP mRNA expression was found in the majority of non–small cell lung cancer (NSCLC) tissues and further diminished levels in metastatic tissues. Reduced expression of both RKIP and E-cadherin was found to promote invasion and metastasis in NSCLC. 102 There is a unique case where a study investigated the effect of phosphorylated RKIP levels and found increased pRKIP was associated with better overall survival (OS) compared to low pRKIP levels in all types of lung cancer patients. 103 The theory suggests that pRKIP in its “inactive” form disables RKIP from regulating pathways conducive to tumor progression. While this is the only clinical study done on pRKIP so far, further studies could uncover cellular details on tumorigenesis and progression.
A particular exception of RKIP regulation can be found in multiple melanoma (MM) tumor cells. It was found that high levels of RKIP were overexpressed in MM cells, specifically overexpressed levels of the Ser153-phosphorylated form of RKIP (pRKIP). This inactivated form is unable to inhibit the MAPK pathway unlike the original RKIP. The overexpression of RKIP and pRKIP in MM cell lines compared to normal cell lines is the first of its nature as usually RKIP is scarcely found in the majority of cancer cells. The dysregulation of the MAPK pathway by the pRKIP form may suggest that pRKIP aids in the phenotypic characteristics of MM cells including drug and apoptosis resistance. The regulation and relationship between RKIP and pRKIP may offer therapeutic treatments and prognostic significance if further studied. 104
While RKIP in melanoma generally acts as a metastatic suppressor,105–107 additional studies need to be conducted to investigate the clinical significance of RKIP in melanoma.
Significantly decreased levels of RKIP are found in nasopharyngeal carcinoma (NPC) compared to its expression in normal nasopharyngeal epithelial tissue. 108 Preclinical studies suggest RKIP to be a statistically important biomarker. 109 All the evidence strongly indicates RKIP as a metastasis suppressor and a marker of poor prognosis.
RKIP expression is significantly reduced in the majority of primary prostate cancer cells11,110,111 and even further diminished in metastatic tissue compared to non-metastatic prostate cancer tissue. 110 RKIP acts as a metastasis suppressor and a regulator of vascular invasion. 11 Further studies demonstrate RKIP’s ability to inhibit STAT3 activation 112 which allows the sensitization of prostate and breast cancer cells to cytotoxic drug-induced cell death. 10
Downregulation of RKIP is associated with a significant number of renal cell carcinoma (RCC) cases and poor prognosis. 113
Depending on the type of thyroid cancer, RKIP expression is varied.114,115 A few preclinical studies indicated decreased RKIP expression to be associated with thyroid cancer progression, while RKIP upregulation was correlated with tumor suppression. 114 Overall, RKIP expression acts as a significant marker of lymph node metastasis. 116
Cancer stem cells and their regulation
Cancer stem cells (CSCs) are tumor initiation cells that are defined by their potential to initiate new tumors, undergo unlimited self-renewal, and proliferate indefinitely to reoccupy the tumor cavity and heterogeneity (multipotency). CSCs share common phenotypes associated with embryonic stem (ES) cells such as self-renewal and high CD44 and low CD24 surface markers. The origins of CSCs are a controversial subject as it is unknown whether they originate from the reversal of differentiation or from the failed early progenitor or stem cells that could not differentiate.117–119 Moreover, it is also debated on whether CSCs follow a hierarchical model where CSCs are similar to normal stem cells versus the dynamic model where the plasticity between CSCs and non-CSCs allows transient reversible states.120–124
CSCs’ associations are linked with two sets of four TFs: one set of Krüppel-like factor 4 (KLF4), SRY (Sex Determining Region Y)-Box 2 (Sox2), Octamer 4 (Oct4), and c-Myc and another set of Oct4, Sox2, Nanog, and Lin28. Studies have shown that both groups are sufficient and essential to induce the signaling events that give rise to characteristics of induced pluripotent stem (iPS) cells in reprogrammed somatic cells by triggering epigenetic chromosomal events including histone acetylation, DNA demethylation, and methylation of promoter regions. 125 Herein, the focus will be on the relationship between RKIP and Oct4, KLF4, Sox2, and Nanog signaling pathways.
Relationship between RKIP and Oct4
As a TF encoded by the POU5F1 gene, Oct4 acts as a positive regulator for other TFs including Sox2 and Nanog. Oct4 specifically is irreplaceable as a component in the four-part set of TFs that maintain the ES cell state as it cannot be replaced by any members from the same protein family.111,126,127 One of the main functions of Oct4 is to uphold the pluripotent cell state.127–129 There may be a connection between the maintenance of pluripotency by certain TFs upregulated in ES cells and CSCs including Oct4 and DNA repair, replication, and stabilization.129,130
As mentioned previously, two different sets of TFs consisting of Myc, Oct4, Sox2, and KLF4 and also Lin28, Oct4, Sox2, and Nanog are grouped together according to their ability to create iPS cells from human fibroblasts, and Oct4 is involved with both groups.12,131,132 In an experiment whereby there was a KO of Oct4 expression, the expressions of Sox2 and Nanog were dramatically reduced which supports the theory that these three TFs work cooperatively to keep the cell in the pluripotent cell state in human embryonic stem cells (hESCs). 128 Among the inducers that activate Oct4 gene expression, FoxD3, Nanog, Lin 28-induced activation, and Oct4 itself act to control the levels of Oct4. In the negative feedback loop, an increase in Oct4 expression would cause Oct4 binding to the Nanog promoter and inhibit the transcription of the Nanog gene and thus reduces Oct4 expression. 133
RKIP ties into the function and regulation of Oct4 by the induction of Lin28, an miRNA (microRNA)-binding protein. Lin28 expression is inhibited by RKIP which in turn upregulates Let-7, an miRNA involved in the activation of the CSC state. 12 On the other side, Lin28 also induces Oct4 expression and may overlook its translational regulation. 134 Thus, RKIP may indirectly downregulate the expression of Oct4 by Lin28. Upregulation and downregulation of Lin28 consequently showed a proportional expression of Oct4 and both were actively expressed in ESC-like state types. The decreased expression of Lin28 and, consequently, Oct4 caused the induction of differentiation, keeping consistent with Oct4’s role as a gatekeeper of pluripotency.135,136 A summary of the interactions can be found in Figure 7. Oct4 is a delicate protein to keep in balance as a 50% reduction or addition of Oct4 levels can dictate the differentiation to embryonic or trophectodermal lineages and influence the maintenance of the normal cell phenotypic state. 137

The downstream signaling pathway in association with RKIP-mediation inhibition of Lin28 and the CSC-related transcription factors: Oct4, Sox2, KLF4, and Nanog.
While RKIP can inhibit the MAPK pathway by binding to Raf-1, which is a target molecule of Ras, an increase in Ras expression was found in overexpressed Stk40 cells. Moreover, Stk40 can induce the activation of ERK by Rcn2 but the exact mechanism is unknown. 138 In addition to the previous mechanisms, there is a possibility of the signaling pathways intersecting between Oct4, leukemia inhibitory factor (LIF)/Stat3, and KLF4, another core TF regulated in pluripotency in CSC-like state cells. Thus, RKIP’s role in the Stat3 pathway could suggest crosstalk pathway mechanisms between RKIP and Oct4, and possibly RKIP with KLF4.111,139–141
Relationship between RKIP and KLF4
KLF4 or the Krüppel-like factor 4 is a zinc finger TF that stunts cell growth by upregulating the transcription of p21 and suppressing cyclin D1. 142 KLF4 enjoys a diverse array of functions including the function as an enhancer of E-cadherin (CDH1) in breast cancer cells 143 and a suppressor of Slug/Snail2 in prostate cancer cells. 144 Its major role is to act as the substrate for the microRNA-7 (miR-7) by binding the miR-7 to two of the possible 3′ untranslated regions of the KLF4 gene. Downregulated KLF4 expression is found to inhibit the metastasis of CSCs in breast cancer cells to the brain but not to the bone. It is also found that the expression of miR-7 is inversely correlated with the expression of KLF4 in which the upregulation of miR-7 resulted in the decreased expression of KLF4. 145 The balance between the various TFs and the epigenetic controls determines the phenotypic expression of CSCs. 125
KLF4 and RKIP do not interact directly with each other but rather through an indirect contact by regulating the Oct4/Sox2/KLF4 complex in the MAPK pathway. The expression of RKIP is associated with the downregulation of Oct4 by the expression of Lin28 and Let-7. To maintain pluripotency in cells, the Oct4 and Sox2 bind together to form a heterodimer before they latch onto different binding sites on the KLF4 C-terminal region. The Oct4/Sox2/KLF4 complex will then bind to the Nanog promoter to transcribe the gene that allows the transition and maintenance of the cellular pluripotency as shown in Figure 8.146–148
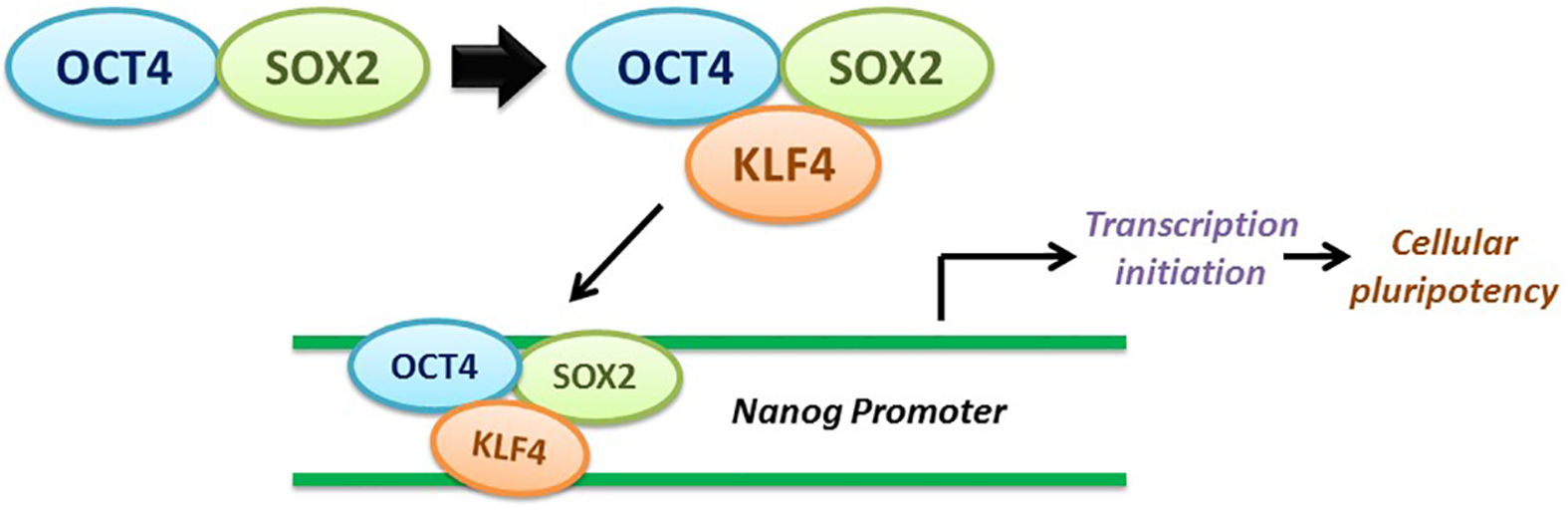
A closer look at the transcription factor complex of Oct4, Sox2, and KLF4. When they bind together, the complex then binds to Nanog promoter to induce the CSC-related transcription factor and promote cellular reprogramming.
In addition, the expression of Let-7 in the presence of Oct4, KLF4, and Nanog promotes the differentiation process of CSCs from the iPS cell state. 149 RKIP is known to function as a metastasis suppressor and an inhibitor of the invasive phenotype. The combination of the expressions of RKIP, Let-7, and KLF4 allows the upregulation of CSC differentiation, the reduction of CSC proliferation, and a decrease of the total frequency of CSC available. Moreover, it is implicated that KLF4 and RKIP interact through the Let-7 group of miRNAs.12,150 The initial step involves the RKIP-mediated inhibition of Lin28 to upregulate Dicer or Drosha processing of Let-7 pre-mRNA. Lin28 is suggested to suppress Dicer processing of Let-7 pre-mRNA. 151 Without processed Let-7 pre-mRNA, there is an increase in Lin28 expression, which in turn upregulates the expression of Oct4 to interact with KLF4 and Sox2 to bind to the Nanog promoter.
Another connection between RKIP and KLF4 is the link between eHSP90 to the MAPK pathway. KLF4 appears to regulate the intracellular chaperone molecule HSP90 which is involved in the proper folding of essential signaling molecules such as Raf-1. 152 There is also an extracellular HSP90 that is suggested to promote cell motility by regulating the matrix metalloprotease (MMP)-2/9 demonstrated in prostate cancer cell lines.
The third link between KLF4 and RKIP could possibly involve p53 and p21 regulation. RKIP is known to induce apoptosis-mediated by p53 and suppress tumor metastasis, while KLF4 upregulates p53-dependent p21 expression.153–155
Relationship between RKIP and Sox2
Sox2 is identified as an oncogenic TF whose regulation is affected by another TF, Oct4. 128 In a study done with mice diagnosed with skin squamous cell carcinoma, Sox2 target genes are involved with tumor invasiveness and proliferation which contribute to the phenotype of tumor-initiating cells and CSCs. 156 Sox2 appears to be an essential component in the de-differentiating process of a cell to return to the iPS state in lung and esophageal squamous cancers, but not crucial for ESC maintenance. One of its main roles is to conduct hESC self-renewal by offsetting the expression of Sox3 with Sox2. 157 If both Sox2 and Sox3 are deleted, self-renewal and cell differentiation abilities are lost. However, the absence of Sox2 led to the expression of Sox3, and Sox3 expression is low in hESCs, but hESC self-renewal ability was intact.
In addition, the posttranslational modifications can greatly impact the activity of Sox2. It was found that the sumoylation of Sox2 hindered the ability to bind to DNA, whereas the sumoylation of Oct4 was found to do the opposite and aid in the DNA-binding affinity. The suppression of acetylation of Sox2 at the lysine 75 residue is found to impede nuclear translocation of Sox2 and continues the hyper-acetylation expression of Sox2 target genes. Moreover, upregulation of Sox2 acetylation decreases the expression of Sox2 by ubiquitination and proteasomal degradation which suggests that acetylation of Sox2 is a major modulator of Sox2 functionality.158–160
RKIP-mediated regulation of Sox2 happens indirectly through the inhibition of Oct4. When RKIP functions to suppress the MAPK pathway, it also inhibits the activation of ERK. Thus, Myc is inhibited which suppresses Lin28 expression by RKIP-mediated upregulation of Let-7, an miRNA that blocks the activation of Lin28. Finally, the cascade of events inhibits the expression of Oct4.12,133,161 Oct4 and Sox2 interact together to form a heterodimer complex to bind to the Nanog promoter along with KLF4 to activate the Nanog gene expression. Remarkably, Sox2 can bind to the Nanog gene even without forming the heterodimer complex with Oct4. 147 This single monomer interaction can regulate ES cell self-renewal by tryptophan and tyrosine residue stackings.
Relationship between RKIP and Nanog
As a TF, Nanog plays a multifaceted role in pluripotency and ES cells by regulating the expression of Oct4, Sox2, KLF4, and itself. 162 Nanog is a 305-amino acid protein that contains a DNA-binding domain on its N terminus, a homeobox domain, and a tryptophan-enriched region on its C terminus.163–166 The regulation of Nanog can occur through several pathways such as the indirect association with the MAPK pathway and the Lin28-Let-7 cascade and the direct connection to the LIF-STAT-3 pathway. Nanog transcriptional expression is regulated by a complex of various CSC-related TFs including the Oct4, Sox2, KLF4, Nanog itself, and others. The Oct4/Sox2 heterodimer binds directly to the Nanog gene to increase its expression. 167 Consequently, the overexpression of Oct4, Sox2, and Nanog is correlated to tumorigenesis. 118 It appears that the delicate balance of these TFs influences the cell fate and the stem cell phenotype. In particular, Nanog is identified as one of the key inducers of the CSC state type due to its implied role in maintaining pluripotency and as an inducer of apoptosis and cellular self-renewal. The coordination of both Oct4 and Nanog has been implicated to be an inducer of the stem cell phenotype as demonstrated by the hepatocellular carcinoma–based study on the stem cell characteristics and the promotion of EMT. 168
RKIP may play an indirect role in the mediation of Nanog expression in CSCs through several pathways. Upregulation of the Raf-1/MEK/ERK pathway can induce the phosphorylation of Nanog at Serine 52 by ERK suggesting that the MAPK pathway may influence pathways associated with the characteristics of self-renewal of CSCs and ES cells. This discovery came about from the effects of the MEK inhibitor PD98059 that causes a decrease in Gata6 expression and an increase in Nanog expression, in which Gata6 is normally regulated by ERK signaling and, thus, ties in the role of the MAPK pathway with Nanog expression. 169
The close relationship between Oct4 and Nanog becomes apparent as RKIP indirectly regulates Oct4 in two related pathways, and consequently also indirectly influences Nanog expression as well. The first is the most significant, where the normal function of RKIP is to inhibit the Raf-1/MEK/ERK signaling cascade, but when RKIP is inhibited, ERK can phosphorylate the TF Myc that can now bind to the Lin28 promoter and upregulates its gene product, which in turn activates Oct4 expression. Oct4 normally forms a heterodimer with Sox2 to bind to the Nanog promoter to induce Nanog expression, but with RKIP inhibition, it can effectively shut off the expression of Oct4 and its downstream targets such as Nanog. 133
Another potential pathway of RKIP-mediated regulation of Nanog is derived from the binding of p53, a tumor suppressor product, to the Nanog promoter in murine ES cells. When p53 responds to DNA damage, it may bind to two consensus binding motifs that induce the inhibition of Nanog in order to repair genetic injury. This pathway is reminiscent of the RKIP cellular senescence regulated by p53, but this connection has yet to be confirmed.118,153
Clinical implications associated with RKIP and CSC TFs
The next step in the investigation of RKIP and CSCs is to search for possible therapeutic treatments to put into clinical use. The hypothesis with regard to CSCs is that their existence supports and drives the malignancy of various tumors. 170 After assessing the commonly overexpressed levels of TFs associated with the CSC phenotype, it is possible that novel therapeutic interventions could take advantage of pluripotency-related pathways to prevent or hinder the growth of CSCs. Novel therapeutic targets could function by inhibiting or downregulating the expression of Oct4, Sox2, KLF4, or factors related to NF-κB.
One study has explored the use of a thioridazine, an antipsychotic drug that inhibits dopamine receptors localized on CSCs and breast cancer cells. It is found to act against only neoplastic cells and leukemia CSCs. When thioridazine was used to treat neoplastic human pluripotent stem cells, the drug was found to reduce Oct4 levels significantly below the control, suggesting that the treatment bypassed the differentiation obstacle. 171 Thioridazine has the potential to explore further therapeutic treatments using dopamine receptors expressed on CSCs as biomarkers for a variety of cancers.
Another study investigated the development of an Oct4-dependent oncolytic adenovirus to specifically target bladder tumors that contain overexpression of Oct4. An oncolytic adenovirus (AdLCY) was developed with regard to regulation by Oct4 and hypoxia, a common feature of surviving cancer cells. AdLCY was found to exhibit antitumor properties against human bladder tumors transplanted in mice. In addition, it could specifically locate and inhibit the growth of CD44- and CD133-positive bladder tumor cells. 172
Studies suggest there may be a correlation between the associated stem cell–like features of Oct4 and Sox2 and the clinical significance on malignant tumors. Oct4 and Sox2 demonstrate clinical importance in cervical cancer, apparent by the correlation between poor prognosis in patients and overexpressed levels of Oct4 and loss of Sox2. 173 However, it is difficult to determine the exact results from Sox2 expression as there are conflicting results of having high expression of Sox2 in lung cancer correlating to low patient survival, whereas there was low expression in esophageal squamous cancer associated with poor prognosis. 174
Additional research has discovered that silencing Sox2 rescues the efficacy of the cancer drug tamoxifen that is used as an endocrine therapy to treat breast cancer. There is growing prevalence of tamoxifen-resistant breast cancer cells that express high levels of Sox2 and also become more difficult to treat. Overexpression of Sox2 in the resistant tumor cells suggests that Sox2 is involved in the Wnt signaling in CSCs. By downregulating Sox2 expression, Wnt signaling decreased as well and thus re-sensitized the resistant cells to tamoxifen. 175
There are pending clinical trials that are pursuing further research on the effect of different therapeutic drugs on the CSCs and their respective malignancies. By understanding the role of CSC-related TFs that help induce pluripotency and tumorigenesis, it becomes possible to selectively target these pathways and produce therapies to treat cancers. The extent to which the therapies can treat cancers through CSC-related pathways is unclear and will be have to be determined through further research and clinical testing.
Conclusion
This review summarizes the potential correlation between RKIP expression and the expression of several CSC TFs. It was found that there were crosstalks between RKIP signaling pathways and several TFs, namely, Oct4, KLF4, Sox2, and Nanog. These crosstalks suggest that RKIP may be implicated in the regulation of the CSC phenotype, though it will need to be validated experimentally.
RKIP inhibits the miRNA binding protein Lin28 whereby it inhibits the induction of Let-7, in part, due to the suppressive role on Raf-1 and Lin28. 12 Another mechanism by which Oct4 and RKIP may interact is via the serine-threonine kinase 40 (STK40). STK40 is inhibited by octamer-binding transcription factor 4 (OCT4) and STK40 stimulates ERK via RCN2. 138
KLF4 does not interact directly with RKIP, but it interacts indirectly via regulation by RKIP of the Oct4/Sox2/KLF4 complex through the MAPK pathway.12,146 The expression of RKIP inversely correlated with the expression of Oct4 via the expression of Lin28 and Let-7. RKIP suppresses the metastatic phenotype via signaling cascades that include MAPK, MEK, Lin28, Let-7, and downstream Let-7 targets such as the chromatin remodeling factor HMGA2, known to induce the activation of TFs involved in EMT such as Snail1.12,150
RKIP upregulates Dicer or Drosha processing of Let-7 pre-mRNA via inhibition of Lin28. Lin28 has been proposed to inhibit Dicer processing of Let-7 pre-mRNA.12,151 The inability of Let-7 pre-mRNA to be processed results in the upregulation of Lin28, which promotes the expression of Oct4, which then interacts with KLF4 and Sox2 to form the complex that binds at the Nanog promoter and enables the transcription of the Nanog gene. Both KLF4 and RKIP regulate NF-κB activity. RKIP inhibits the NF-κB pathway.38,176 KLF4 interacts with the p65 subunit of NF-κB.154,177
RKIP inhibits Sox2 via its inhibition of the MAPK pathway. The activation of ERK leads to the activation of MEK and promotes Lin28 and leads to the expression of Oct4.12,133,161 RKIP regulates the expression of Let-7 miRNA that suppresses Lin28. Lin28 targets and promotes the expression of Oct4, which forms a heterodimer with Sox2. 147 Therefore, its most significant interaction of RKIP with Sox2 is via its regulation with Oct4.
It was reported that Nanog is phosphorylated by phospho-ERK at serine 52 and results in the negative regulation of Nanog transactivation. 178 This inhibition by RKIP is mediated by inhibiting phospho-ERK. Also, Oct4 is required for Nanog expression and Oct4 is indirectly regulated by RKIP via inhibition of Lin28-mediated expression of Oct4 and failure of the formation of the heterodimer complex of Oct4 and Sox2. In addition, another link between RKIP and Nanog is via p53. p53 binds to the Nanog promoter and suppresses its transactivation, and RKIP-p53 mediates cellular senescence.117,153
Clearly, if these above crosstalks exist, it would suggest that the induction of RKIP would function as a metastasis suppressor and as a chemo-immunosensitizing agent in CSCs. New agents have to be developed that induce the expression of RKIP which would subsequently result in directly inhibiting the TFs that regulate the CSC phenotype as well as sensitizing the whole tumor to various chemo-immunotherapeutic modalities. In addition, based on the crosstalks between RKIP and TF pathways, it would also be useful to determine the interrelationship between the active non-phosphorylated form of RKIP and the inactive phosphorylated form of RKIP with respect to their roles and the regulation of the CSC phenotype. Overall, this review has postulated a new paradigm that needs to be investigated experimentally and validated for its translational application in patients with cancer.
Footnotes
Acknowledgements
The authors acknowledge the research support for the reported publications used in this review and include the Jonsson Comprehensive Cancer Center at UCLA; the University of California Gene Medicine Program; the UCLA AIDS Institute; and the NCI-RO1-CA133479. They acknowledge the support of Ailinia Lao and Arah Cho who designed the figures, and they also thank Samantha Kaufhold for her assistance in editing the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.