
Abstract
c-Met (mesenchymal–epithelial transition factor) is a tyrosine kinase receptor activated by hepatocyte growth factor and regulates multiple biological processes, such as cell scattering, survival, and proliferation. Aberrant c-Met signaling has been implicated in a variety of cancer types, including colorectal cancer. c-Met is genetically altered through various mechanisms that is associated with colorectal cancer progression and metastasis. Especially, in colorectal cancer, preclinical evidence for the aberrant activation of the c-Met signaling exists. Accordingly, molecular targeting of c-Met receptor could be a promising strategy, in the treatment of colorectal cancer patients. Recently, it was also shown that crosstalk between c-Met and other cell surface receptors attributes to tumorigenesis and development of therapeutic resistance. Characterization of the molecular mechanisms through which c-Met crosstalks with other receptors in favor of tumor formation and progression remains to explore. This review will describe the mechanisms of aberrant c-Met signaling in colorectal cancer and discuss on additional roles for c-Met receptor through crosstalk with other tyrosine kinase receptors and cell surface proteins in colorectal cancer. Novel therapeutic approaches for c-Met pathway targeting will also be discussed.
Introduction
Colorectal cancer (CRC) is one of the most prevalent cancers, and 1.2 million new cases are diagnosed worldwide annually.1,2 CRC is the second reason of cancer-related deaths worldwide. 3 Despite several advancements regarding the diagnosis and treatment of CRC, approximately 30% of CRC patients develop distant metastases after curative surgery or following treatment with adjuvant chemotherapy and/or radiotherapy. 4 Recent investigations have found many clinical and pathological biomarkers that are associated with the prognosis of CRC. Identification of critical receptors and signaling pathways is crucial for development of efficient targeted therapies in patients with CRC.5,6 c-Met (mesenchymal–epithelial transition factor) is a cell surface receptor tyrosine kinase (RTK) for hepatocyte growth factor (HGF)/scatter factor (SF).7,8 c-Met/HGF activation initiates a variety of cellular responses such as motility, proliferation, wound healing, tissue regeneration, epithelial to mesenchymal transition (EMT), and angiogenesis. 9 c-Met receptor regulates many processes in the development and progression of cancer cells, particularly regulation of tumor invasion, metastasis, and angiogenesis. 10 In the context of CRC, c-Met overexpression and activation are associated with progression and metastasis. 11 Moreover, several crosstalks have been reported between c-Met and other growth factor receptors in CRC. These crosstalks result in tumor progression and may play a crucial role in acquired resistance to targeted therapy. 4 The aim of this study is to summarize the available preclinical and clinical evidences of aberrant c-Met signaling in CRC and understanding the relevant mechanisms underlying in CRC formation, progression, and resistance to targeted therapies.
Molecular structure and biological function
c-Met, RTK, is a 190-kDa heterodimer glycoprotein consisting of an extracellular 50-kDa α-chain and a transmembrane (TM) 145-kDa β-chain. The extracellular α-chain is linked to the TM β-chain by a disulfide bond. 12 The extracellular chain of c-Met includes the semaphorin (SEMA) domain, the plexin–semaphorin–integrin (PSI) domain (because it is present in plexin, SEMA, and integrin cysteine-rich), and four immunoglobulin-like plexin transcription (IPT) domains (immunoglobulin-like fold shared by plexins and transcription factors). Ser975 and Tyr1003 residues in the juxtamembrane domain are important in regulation of c-Met. 13 The catalytic domain of c-Met includes two tyrosine residues (Tyr-1234 and Tyr-1235) that positively regulate the kinase activity. The multifunctional carboxy-terminal docking site is responsible for the recruitment of various intracellular signaling and adaptor proteins. 12 c-Met is expressed mainly by epithelial cells and to a lesser extent by vascular and lymphatic endothelial cells, 14 as well as neural cells, hematopoietic cells, hepatocytes, 15 and perycites. 16 c-Met binds to its physiological ligand, HGF/SF. 17 HGF/SF is a multifunctional growth factor. It is secreted as a single-chain inactive protein and subsequently activated by extracellular proteases.11,18 Mature HGF consists of a disulfide-bond heterodimer of the α-chain subunit (69 kDa) and a β-chain subunit (34 kDa). The α-chain composes of an N-terminal hairpin domain and four kringle domains, while the β-chain contains a serine protease–like structure that lacks catalytic activity. 19 On the molecular level, binding of HGF to c-Met leads to its activation by autophosphorylation of Tyr 1234 and Tyr 1235 residues in intercellular tyrosine kinase (TK) domain. 20 Phosphorylation of Tyr 1349 and Tyr 1356 residues in the docking site recruits intracellular adaptor molecules such as Gab1 (growth factor receptor bound protein 2–associated binder 1), Grb2 (growth factor receptor–bound protein 2), Src, and SHC. 21 HGF/c-Met binding triggers several signal transduction pathways such as Rac1/Cdc42 and the Src/focal adhesion kinase (FAK), the phosphoinositide 3-kinase (PI3K)/Akt, and the Ras/MEK. 22 These phenomena play an important role in cell proliferation, survival, motility, migration, invasion, angiogenesis, and branching morphogenesis. 23 c-Met receptor is negatively regulated by an E3 ubiquitin ligase, Cbl. Following c-Met activation, Cbl recognizes and binds the phosphorylated Tyr1003 residue in the juxtamembrane domain. 24 Cbl binding induces polyubiquitination and degradation of c-Met in late endosomal or lysosomal compartment through a proteasome-dependent manner. 25
Aberrant c-Met signaling in CRC
The HGF receptor, which is often aberrantly activated in human cancers, is an interesting candidate for targeted therapy. 26 The oncogenic dysregulation of the HGF/c-MET pathway has been implicated in a wide range of human epithelial cancers including liver, lung, colorectal, breast, pancreatic, ovarian, prostate, hepatic, and gastric cancers.27–31 This pathway results in cell survival and migration and tumor development and progression.27,32 c-Met over-activation occurs via several molecular mechanisms, including gene amplification, 31 overexpression, 33 mutations, 34 or RTK transactivation or changes in ligand-induced autocrine or paracrine signaling; 35 all of them have been reported in several human tumor types. 11 c-Met overexpression is reported in many human cancer cells, such as hepatocellular carcinoma (HCC), non-small-cell lung cancer (NSCLC), and approximately 50% of advanced gastric cancers that have been associated with poor prognosis.36,37 Overexpression of c-Met has been reported in 30%–70% of CRC tumors.38,39 In CRC, the expression of c-Met at messenger RNA (mRNA) and protein levels have been detected in 30%–91% of samples when assayed by northern blot and reverse transcription polymerase chain reaction (RT-PCR) and 57%–100% by western blot and immunohistochemistry (IHC) analyses.2,38 Several studies have demonstrated a significant overexpression of c-Met mRNA and protein in CRC tumors compared with adjacent normal colon mucosa. For example, Zeng et al. 39 reported that 69% of primary CRCs demonstrated c-Met protein overexpression.
The major reason of CRC-related death is metastases. c-Met overexpression has been found to be associated with tumor progression and metastasis of CRC.3,40 c-Met expression is reported in more than 50% of colorectal lesions of dysplastic adenoma and invasive carcinoma cases. 41 Furthermore, protein levels of c-Met were remarkably increased in advanced stages (stages III and IV) compared with early stages (stages I and II) of CRC. The IHC analysis revealed that expression of c-Met in endothelial cells of blood vessel is associated with progression of human CRC.2,39 Most recently, a meta-analysis study indicated a remarkable association between c-Met overexpression and poor overall survival (OS) and progression-free survival (PFS) in CRC. c-Met gain of function mutations is rare in cancers but may associate with tumor development.3,4 c-Met point mutations have been found in a wide range of solid tumors, and it is presumed that a number of mutations could lead to constitutive activation of c-Met. The frequency of missense mutations in c-MET TK domains are enriched in metastatic lesions from head and neck squamous cell carcinoma (HNSCC), 42 renal cell carcinoma (RCC), 43 childhood HCC, 44 and colorectal carcinoma. 26 Denaturing high-performance liquid chromatography (DHPLC) or cold single-strand conformation polymorphism (SSCP) analyses in primary gastric cancer showed that missense mutations in c-MET juxtamembrane domain were detected in only 1% of patients. 45 Somatic mutations have been detected in juxtamembrane domain of c-Met, resulting in the loss of Cbl E3-ligase binding and reducing c-Met ubiquitination and degradation. Moreover, somatic mutations were correlated with c-Met overexpression in primary tumors and were reported in about 3% of NSCLC patients.46,47 Other mutations can also be found in the c-Met SEMA domain in lung cancer, which result in the HGF binding and receptor dimerization. 4 Ligand-dependent c-Met activation has been implicated in the majority of solid gastrointestinal tumors, particularly in CRC. 48 Overexpression of c-Met and HGF in CRC microenvironment is associated with poor clinical outcome.49,50 In summary, there is a pile of evidences showing c-Met autocrine and paracrine activation, c-Met overexpression, and mutational activation in human cancer cells suggesting that HGF-c-Met targeting could be a promising approach in cancer therapy.
Crosstalk with other RTKs
Crosstalk among c-Met signaling pathway and other RTKs has been substantially studied because of its potential importance in understanding the mechanisms of resistance to targeted therapies (Figure 1). Numerous evidences suggest that crosstalk between abnormal RTKs can significantly influence efficacy of TK inhibitors. Here, some RTK crosstalks with HGF/c-Met pathway, including the vascular endothelial growth factor receptor (VEGFR), the epidermal growth factor receptor (EGFR), the recepteur d’origine nantais (RON), and the insulin-like growth factor receptor (IGFR), as well as metastasis-associated in colon cancer 1 (MACC1) are described.

c-Met signaling pathways and their crosstalk with other membrane receptors. c-Met signaling can be modulated by crosstalk with other membrane receptors. HGFR, hepatocyte growth factor receptor; VEGFR, vascular endothelial growth factor receptor; EGFR, epidermal growth factor receptor; IGF1R, insulin-like growth factor 1 receptor; SOS, Son of Sevenless guanine nucleotide exchange factor; GRB2, growth factor receptor–bound protein 2; ERK, extracellular signal–regulated kinase; PI3K, phosphoinositol 3-kinase; Akt, protein kinase B; mTOR mammalian target of rapamycin; FAK, focal adhesion kinase; CRK, CT10 sarcoma oncogene cellular homolog; STAT, signal transducer and activator of transcription factor.
c-Met and VEGF
The HGF/c-Met pathway activates multiple signaling cascades including SRC/FAK, STAT3, PI3K/AKT, and RAS which contribute in angiogenesis, directly by stimulating endothelial cells growth or indirectly by elevating the expression of angiogenic factors such as VEGF and suppressing the activity of thrombospondin1—a negative mediator of angiogenesis. 51 Inhibitors of HGF/c-Met signaling pathway have been demonstrated to abolish angiogenesis in multiple in vitro and in vivo assays. A synergistic activity of HGF and VEGF is found in endothelial cells.52,53 Matsumura et al. 54 found HGF/c-Met signaling pathway as regulator of VEGF expression in CT26 VEGF overexpressed cells. Recent studies demonstrated that hypoxia, results from VEGF pathway inhibitors, induces c-Met expression which leads to resistance to VEGF inhibition. 32 Elevated expression of c-Met was observed following sorafenib treatment (a VEGFR inhibitor approved for treatment of HCC) which is correlated with cell migration, invasiveness, and short PFS.
Tivantinib (a c-Met pathway inhibitor) and sorafenib combinational therapy was shown to increase cytotoxicity in HCC cells.55,56 Another study revealed that HGF overexpression causes resistance to lenvatinib (a VEGFR inhibitor). This resistance is overcome by golvatinib (a c-Met inhibitor).57,58 It is also reported that HGF confers resistance to sunitinib, an inhibitor of multiple kinases, including VEGFR2, by compensating for inhibited angiogenesis.59,60 Previous studies showed that activity of the c-HGF-Met/Gab1 signaling pathway leads to VEGF production in EGFR-mutant lung cancer cell lines. 61 Given that targeting VEGF signaling is critical in anti-angiogenic therapy, targeting c-Met pathway in combination with VEGF pathway inhibitors, is of major priority for efficient anti-angiogenic therapy.
T-1840383, a small-molecule kinase inhibitor with capability of targeting both c-Met and VEGFRs, has been demonstrated to successfully suppress constitutively activated c-Met phosphorylation and VEGF-induced VEGFR2 phosphorylation. T-1840383 efficiently inhibited angiogenesis and proliferation in various human tumor xenograft mouse models. 62
Cabozantinib, another small-molecule inhibitor of c-Met and VEGFR2, is an example of combined targeting of VEGF/c-Met signaling which is recently approved by Food and Drug Administration (FDA) for progressive, metastatic thyroid cancer and demonstrated antitumor activity in preclinical models of various malignancies including lung, glioma, pancreas, thyroid, prostate, and CRCs. In breast cancer models, cabozantinib remarkably declined the growth of cancer cells compared to sunitinib. 32
Mezquita et al. 63 showed that phosphorylation of c-Met in LoVo colon cancer cell, oxaliplatin-resistant cell (LoVoR) correlates with the low expression of VEGF, while addition of recombinant VEGF into the culture attributes to the decline of c-Met activation, confirming c-Met activation as a compensatory mechanism of anti-VEGF therapy.
Taking these findings and variable resistant phenotypes in various CRC cell lines into account, c-Met and VEGF co-targeting in colorectal tumors is critical to achieve prolong survival and overcome resistance.
c-Met and RON receptors
RON and c-Met belong to the SEMA family of TM RTKs and share many structural similarities. c-Met and RON with 63% homology in their extracellular domain and 25% homology within the TK domain possess very similar functional domains. Both receptors have extracellular domain composed of two chains of α and β, the SEMA as ligand-binding domain, PSI, the IPT, TM, and an intracellular TK domains. 64 c-Met and RON contribute to embryonic development, and their overexpressions or aberrant activations are reported in various types of cancers. Numerous evidences indicate contribution of these RTKs’ signaling in tumor progression by controlling cellular proliferation, motility, angiogenesis, maintenance of cancer stem cells, and protection from apoptosis.64,65 Overexpression of RON in primary tumors such as colon and breast cancers is predictive of patient survival and correlates with clinical and pathological parameters. 66 Moreover, Han et al. 67 revealed that tumor cell survival and progression in gliomas, multiple myeloma, prostate, colon, breast, lung, and pancreas cancers rely on HGF and macrophage-stimulating protein (MSP) expressions and functions. 68 c-Met/RON activation is documented to contribute in malignancy. Overexpression of c-Met/RON promotes EMT, a unique feature of cancer stem cells. RON and Met concomitant overexpression is detected in many tumor types.65,68 Lee et al. 66 found that overexpression of RON and c-Met in CRC is associated with a remarkably poor clinical outcome and short-term disease-free survival. Various mechanisms underlay on abnormal expression and activation of c-Met and RON such as gene amplification, enhanced TK activity due to point mutations, and aberrant splicing of RON.65,69 c-Met and activated RON can homo- or heterodimerize with each other to trigger phosphorylation of TK domain. Activation of downstream signaling cascades including PI3K/AKT and Ras/MAPK occurs following the recruitment of adaptor proteins.
They may interact with and modulate signaling of other RTKs. For example, c-Met and RON signaling promotes angiogenesis through crosstalk with VEGF driven by hypoxia-inducible factor 1-alpha (HIF-1α). c-Met and RON signaling pathways mostly rely on activation of PI3K and MAPK as key adaptor proteins which result in PI3K/AKT, Src, STAT3, NF-κB, FAK, and β-catenin activation, leading to critical cellular responses including cytoskeletal changes, EMT, stemness, invasion, resistance to apoptosis, angiogenesis, and proliferation. 64 Benvenuti et al. 65 by analyzing four different tumor cell lines including SW620 (colorectal adenocarcinoma) showed constitutive expression of c-Met and activation of RON due to specifically transphosphorylation by c-Met. Several studies proved that single targeting of c-Met or RON did not bring about durable inhibition of tumor progression because of transactivation of another RTKs. 68 Several small-molecule kinase inhibitors and neutralizing antibodies targeting c-Met and RON are recently approved by FDA or entered clinical trials.
c-Met and EGFR
Concomitant overexpression of EGFR and c-Met is commonly observed in the same tumors. 50 c-Met has been reported as the first identified factor of resistance to EGFR inhibitors in several cancer types. 70
Numerous studies described EGFR and c-Met crosstalk in which either RTKs can compensate the role of other one in the signaling cascade events.71,72 Bardelli et al. 73 reported acquired resistance to cetuximab or panitumumab in metastatic CRC patients due to amplification of c-Met without any genetic mutations associated with resistance to anti-EGFR therapy (KRAS, BRAF, NRAS, PIK3CA, and HER2). Other studies revealed that tumor cells treated with c-Met inhibitors also develop resistance through sustained high MAPK and PI3K/AKT activity, mediated by EGFR family members.71,74 HGF induces c-Met/PI3K/AKT signaling pathway, leading to gefitinib (an EGFR inhibitor) resistance in lung cancer cells with EGFR-activating mutations. 75 Overexpression of transforming growth factor alpha (TGF-α) following EGFR–c-Met interaction conferred resistance to cetuximab in CRC cells. 76 c-Met activation in the presence of TGFα or EGF (EGFR ligands) and absence of HGF (c-Met ligand) represents a compensatory mechanism by which c-Met induces proliferative/anti-apoptotic MEK-ERK and PI3K-AKT pathways and attributes to resistance to EGFR inhibitors. 77 Simultaneous administration of NK4 (an anti-HGF antibody) and EGFR tyrosine kinase inhibitors (TKIs), as an example of combined targeting of both receptors, remarkably diminished HGF-induced resistance both in vitro and in vivo.70,78 The molecular mechanisms underlying the crosstalk between EGFR and c-Met are investigated in several studies. Downstream proteins including Src, MAPK, and β1 integrins hypothetically affect EGFR-mediated phosphorylation of c-Met. 77 On the other hand, HGF could also induce EGFR phosphorylation, indicating correlative regulation of both receptors.78–82 EGFR modulates oncogenic function of c-Met by inducing c-Met ectodomain shedding. Soluble c-Met (s-c-Met) generated via c-Met ectodomain shedding is associated with malignancy potential in preclinical models. 83 Nath et al. 84 showed that EGFR activation, via EGF or G-protein-coupled receptor (GPCR) agonist and lysophosphatidic acid (LPA), promotes the cleavage of c-Met through activation of the Erk-MAP kinase signaling cascade.
Tanizaki et al. revealed that different heterodimerization patterns between c-Met and other RTKs result in various biological outcomes. They found that based on differential partnerships, c-Met can trigger particular signaling cascades. EGFR and HER3 heterodimerization with c-Met results in cell proliferation and survival. Moreover, RET/c-Met and HER2/c-Met crosstalks play a role in proliferation, survival, and cell migration events. 85 Moreover, c-Met and EGFR are both introduced as CRC stem cell markers. Combined targeting of EGFR and c-Met for the treatment of RAS-wild-type (RASwt) tumors was shown to be more efficient in elimination of cancer stem cells. 86 The latter approach resulted in omission of stemness marker and increased tumor differentiation in preclinical models. 86 Combinational EGFR and c-Met therapy promotes cancer stem cell eradication and reliable tumor regression in CRC. 70 Jun et al.’s investigations in glioblastoma manifested key role for c-Met in sustaining glioblastoma stem cells (GSC) populations. They showed that inhibition of EGFR in a genetically engineered mouse model of glioblastoma multiforme (GBM) with gefitinib resulted in c-Met expression in a subset of cells that have GSC characteristics. 70 Accumulating findings demonstrate that EGFR/Met crosstalk is complex and multi-factorial. Therefore, considering precise crosstalk among c-Met and other RTKs seems necessary in selecting efficient and durable therapeutic strategies.
c-Met and IGF1R
TK receptor, IGF1R, triggers the RAS/RAF/MAPK and the PI3K/AKT/mTOR pathways following activation. 77 c-Met and IGF1R are both considered as mediators of tumor cell migration and invasion. Recently, a substantial crosstalk between HGF/c-Met and IGF1/IGF1R is described. Bauer et al. found that IGF1R and c-Met contribute in migration and invasion of human CRC cells by regulating urokinase plasminogen activator. They also showed that c-Met downregulation entirely suppresses IGF-I-mediated migration and invasion. 87 Varkaris et al. also confirmed essential role of c-Met for migratory effects of activated IGF1R. IGF1R signaling has been shown to participate in ligand-independent activation of c-Met. Induction of c-Met activity, in the absence of its ligand, following direct injection of IGF-1 was reported in a PC3 xenograft model. Delayed IGF-1-induced c-Met activation in multiple cell lines expressing both receptors was confirmed. Src kinase has been described as main player in ligand-independent c-Met activation. 88
Lee et al. describing differential signaling pathway activation in various CRC subclasses demonstrated c-Met and IGF1R over-activation and is associated with poor survival in metastatic CRCs, relating these two RTKs. 89 c-Met and IGF1R expressions are both considered as reasons of resistance to EGFR inhibitors such as cetuximab in metastatic CRC patients. 90
c-Met and MACC1
MACC1 overexpression is reported to be associated with malignancy in colon, liver, and lung cancers. It is also known as a central regulator of the metastatic events induced by HGF/c-Met pathway in CRC. MACC1 and c-Met are considered as prognostic indicators for colon cancer metastasis.91,92 Functional correlation between these two genes is also confirmed in gastric cancer cell lines. 93
Arlt et al. found a prominent positive correlation between MACC1 and c-Met. MACC1 reported to be a major regulator of met transcription. It can induce proliferation, invasion, and metastasis in xenograft cancer models. Small interfering RNA (SiRNA) targeted to c-Met or MACC1 reversed all these properties in vitro and in vivo. This is in accordance with previous findings regarding c-Met siRNA capability in suppression of tumor growth and metastasis. 91
MACC1-dependent biological effects are assumed to be in part via the HGF/Met pathway. In brief, following HGF exposure, nuclear translocation of MACC1 and its binding to c-Met promoter lead to transcriptional activation of c-Met. The latter represents a positive feedback loop by which increased amounts of c-Met protein might bind to more HGF molecules, leading to enhanced HGF/c-Met signaling, cell migration, proliferation, and metastasis. 94 Furthermore, MACC1 seems to be a target of the MAPK signaling pathway. 95
Migliore et al. showed miR-1 as negative regulator of c-Met expression in CRC. They found that miR-1 downregulation in the majority of colon cancers, especially in metastatic tumors, is concomitant with increased level of c-Met expression. Accordingly, the exogenous expression of miR-1 in colon cancer cells resulted in decreased c-Met levels and c-Met-mediated invasion. Finally, concomitant miR-1 decrease and MACC1 increase functionally synergize in promoting c-Met overexpression and can contribute to the progression of colon cancer toward metastases. 96
MACC1 and c-Met protein expressions in early-stage and advanced CRCs and their correlation with clinicopathologic parameters were assessed. These findings suggest that MACC1 expression may contribute to the invasive growth of early-stage CRCs. However, no association between MACC1 and c-Met was reported in the CRC initiation and progression. However, in advanced metastatic CRC patients, upregulation of both MACC1 and c-Met was reported. 92 Given that survival of patients with CRC critically depends on early diagnosis, recognizing new reliable biomarkers is of pivotal importance to promote the prognostic and predictive factors for efficient treatment.
Consequently, many of the c-Met interactions may cause c-Met phosphorylation and activation. The molecular mechanisms underlying these crosstalks need to be further investigated. Nonetheless, these data indicate that c-Met receptor can interact with other receptors and proteins and triggering downstream signaling mechanisms leading to tumor formation, progression, and resistance to antitumor therapies. Multi-targeted therapeutic strategy might be used to address the common problems of crosstalk among signaling transduction mechanisms and thus development of resistance to targeted cancer therapy.
HGF and c-Met inhibitors for treatment of CRC
Several strategies have been introduced for antagonizing HGF/c-Met signaling in CRC. Antibodies can be administered to bind to both c-Met and HGF.97,98 In addition, many approved agents are designed to block specific signaling pathways of tumor formation, survival, proliferation, progression, dissemination, and/or angiogenesis. 99 These agents represent different pharmaceutical indexes such as potency, selectivity, pharmacokinetic (PK) properties and toxicity profiles. 8 A number of well-studied HGF/c-Met inhibitors are presented in Table 1.
Clinical trial for c-Met inhibitors.
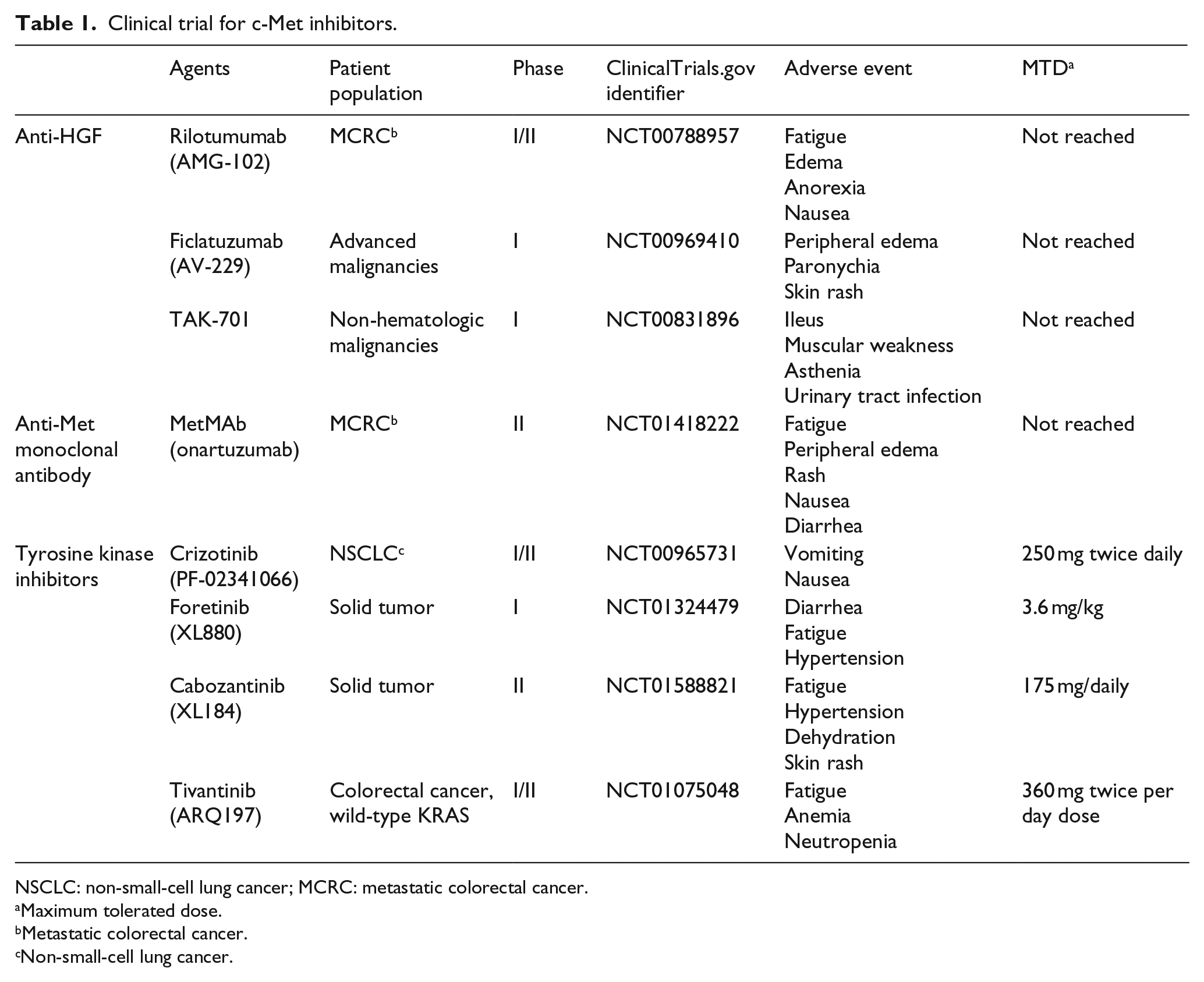
NSCLC: non-small-cell lung cancer; MCRC: metastatic colorectal cancer.
Maximum tolerated dose.
Metastatic colorectal cancer.
Non-small-cell lung cancer.
Anti-HGF monoclonal antibodies
AMG 102 (rilotumumab) is a humanized IgG2 monoclonal antibody, directed against HGF, which hinders interaction between HGF and c-Met.77,100 A randomized phase Ib/II clinical trial of panitumumab combined with rilotumumab or placebo showed significant clinical outcome in patients with KRAS wild-type, advanced CRC. At present, there is no indication of further investigation of rilotumumab in advanced CRC. However, a more precise selection of c-Met positive tumors and the evaluation of rilotumumab as a single factor in both KRAS wild-type and mutated tumors could make the therapeutic index of this agent more effective. Furthermore, rilotumumab continues to be evaluated in other malignancies, attaining phase III development in gastric cancer.77,101
Anti-Met monoclonal antibodies
TK inhibitors
TKIs are small molecules that target adenosine triphosphate (ATP) binding site in the TK domain of RTKs, inhibiting receptor transactivation and enlistment of downstream effectors.
77
TKIs can be separated into two groups based on their selectivity for c-Met.
The variation among these inhibitors seems to mainly reflect their potency and selectivity against these individual kinases. Finally, INCB028060, E7050, JNJ-38877605, and BMS-777607 are dominant, and selective inhibitors of c-Met RTK were first accomplished in human research.8,77
c-Met targeting by microRNAs
MicroRNAs (miRNAs) are small non-coding RNAs that have appeared as key regulators of gene expression by RNA interference. Some miRNAs have been recognized that target c-Met oncogenes such as miR-34a, miR-206, miR-199, and miR-1 that could compete in therapies for silencing c-Met. 98 Recent findings demonstrate that miR-1 is downregulated in CRC with respect to normal tissues, and it has been shown that this miRNA can downregulate c-Met expression in CRC models. Furthermore, re-expression of miR-1 in CRC cell lines causes c-Met-driven decrement in cell proliferation and motility, suggesting that miR-1 can be a feasible candidate for clinical trials of c-Met inhibitors in the therapy of metastatic CRC.128,129
Conclusion
Various preclinical and clinical findings have shown substantial role of c-Met/HGF signaling in solid tumors especially in CRC, and there are convincing evidences from in vitro and in vivo studies that this is a critical pathway in CRC formation and progression. Recently, clinical evidences have confirmed that c-Met is a key oncogene involved in CRC and highlights the therapeutic potential of c-Met inhibitors in CRC. The results of continuous and future clinical trials of anti-c-Met therapy will be prevised, but studies about receptor crosstalk and resistance may need to be clarified in order to increase treatment efficacy. In this review, we have summarized the role of HGF-c-Met signaling pathways in the CRC development and progression. Further investigations are needed to focus on identification of treatment resistance mechanisms and strategies of combination therapy in order to maximize treatment efficacy in patients with CRC.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.