
Abstract
Background
Osteoporosis is a common condition among the elderly, characterized by reduced bone density and an increased susceptibility to fractures. The efficacy of Cinnamomum cassia in treating osteoporosis is recognized, though its precise molecular mechanisms remain unclear.
Objectives
To investigate the molecular mechanisms underlying the therapeutic effects of C. cassia using network pharmacology, molecular docking technology (MDT), and molecular dynamics simulation (MDS).
Materials and Methods
Bioinformatics databases identified active compounds and disease targets. Protein-protein interaction (PPI) networks were constructed, followed by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses. MDT and MDS validated the binding affinity between C. cassia’s active compounds and key targets.
Results
Ten active compounds of C. cassia were identified, modulating the PPAR, HIF-1, AMPK, and cAMP signaling pathways. Key genes include PPARG, PTGS2, PPARA, BDNF, and RXRA. Molecular docking and simulations confirmed high binding affinity between active compounds and targets, supporting their role in regulating bone metabolism.
Conclusion
This study highlights the multi-target mechanisms of C. cassia in treating osteoporosis, emphasizing its therapeutic potential through pathway-based modulation of key genes. These findings provide a basis for further research in modernizing traditional Chinese medicine for osteoporosis management.
Keywords
Introduction
Network pharmacology (Hopkins, 2008) emphasizes multi-target drug discovery (Nogales et al., 2022), integrating systems biology (Jiashuo et al., 2022; Zhao et al., 2023) to enhance drug design and disease management (Tsuchikama et al., 2024). It has provided novel methodologies and fresh perspectives to contemporary medicine, particularly in navigating the intricate terrain of complex drug investigations (Munnangi et al., 2023). Studies indicate that Cinnamomum cassia regulates lipid metabolism and bone remodeling, enhancing osteoblast function and inhibiting osteoclast activity (Dong et al., 2021). These networks represent a pioneering avenue for the design of multi-target drugs and stand as a pivotal facet in the modernization of traditional Chinese medicine (Nogales et al., 2022; Zhang et al., 2023).
This article, rooted in the therapeutic properties of C. cassia and firmly grounded in the principles of network pharmacology (Kim et al., 2023; Lim et al., 2022; Zhang et al., 2019), embarks on an incipient exploration into the mechanisms underpinning the efficacy of this traditional Chinese medicine in the treatment of osteoporosis (Armas & Recker, 2012; Miller, 2016). It painstakingly constructs a comprehensive network of interactions encompassing the active constituents of C. cassia (Lee et al., 2002; Liu et al., 2018), its pharmacological targets, and the genes associated with osteoporosis (Li et al., 2024; Liu et al., 2023; Zhou et al., 2023). This innovative approach challenges the conventional paradigm entrenched in pharmacological thought, one that has traditionally adhered to the concept of “single component, single target, single disease” (Barabási et al., 2011; Li, Liu et al., 2023). The research flowchart is depicted in Figure 1.
The Research Flowchart.
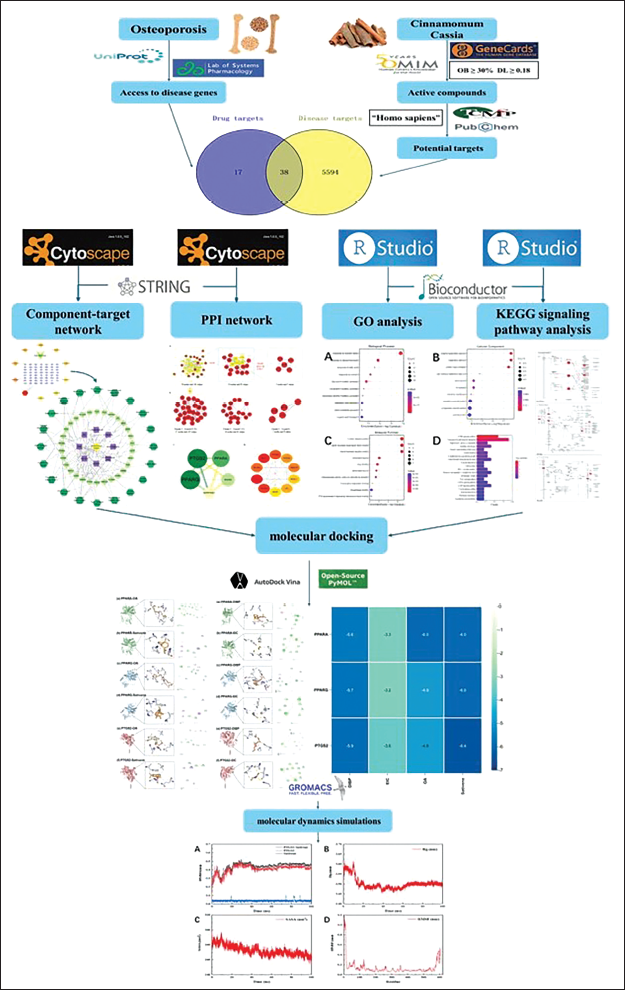
Materials and Methods
Collection of Chemical Constituents and Targets
Chemical constituents of C. cassia were identified using TCMSP (
Prediction of Disease Targets
Intersection targets between C. cassia compounds and osteoporosis were identified using Venny software (
“Drug–Target” Network Analysis
Network topology analysis was conducted using Cytoscape 3.9.1 (Shannon et al., 2003) to visualize drug–target interactions (Shang et al., 2023).
Protein–Protein Interaction (PPI) Network Construction and Analysis
PPI networks (Palukuri & Marcotte, 2021) were constructed using STRING (
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis
GO and KEGG (Kanehisa & Goto, 2000) pathway enrichment analyses were conducted using DAVID (Ding et al., 2024), with results filtered at a p value of <0.05.
Molecular Docking Technology (MDT)
Molecular docking involved ligands such as diisobutyl phthalate (DIBP, PubChem CID: 6782), ethyl isocyanate (EIC, PubChem CID: 8022), oleic acid (OA, PubChem CID: 445639), and sativene (PubChem CID: 11830550). The receptor proteins included PPARA (Uniprot ID: Q07869), PPARG (Uniprot ID: P37231), and PTGS2 (Uniprot ID: P35354). Molecular docking analyses were conducted utilizing AutoDock Vina version 1.1.2 (MDT: Molecular Docking Technology). Three-dimensional protein structures were retrieved from the UniProt database (
The docking box for the PPARA protein was configured with dimensions of 100 Å × 74 Å × 126 Å, with a grid spacing of 0.786 Å, and the coordinates were –4.448, –4.251, and –7.491. For PPARG, the docking box measured 74 Å × 64 Å × 116 Å with a grid spacing of 0.983 Å, and the coordinates were –5.943, –5.943, –5.943, and –5.983. The PTGS2 protein’s docking box dimensions were 72 Å × 82 Å × 68 Å, with a lattice spacing of 0.967 Å, and coordinates at –3.391, 1.053, and –3.585. The conformation with the lowest binding energy was determined to represent the most plausible binding mode between the ligand and the protein. Visualization of the ligand–protein interactions was carried out using PyMOL 2.4.
Molecular Dynamics Simulation (MDS)
MDSs were performed utilizing GROMACS version 2019.6 (Berendsen et al., 1995; Wu et al., 2023) to study protein–ligand interactions. The simulations used amber99sb-ildn and GAFF force fields for proteins and ligands, respectively, for the generation of parameters and topologies. The simulation box was configured so that each protein atom maintained a minimum distance of 1.0 nm from the box edges. The box was filled with SPC216 water molecules as an explicit solvent, Na+ and Cl– counterions were introduced to replace water molecules, ensuring electrical neutrality within the system. To minimize unreasonable atomic contacts or overlaps, the optimization of the system was performed using the steepest descent method. Subsequently, pre-equilibration was conducted with 100 ps of the NVT ensemble at 300 K, followed by 100 ps of the NPT ensemble at 1 bar. Under periodic boundary conditions, the main MDS was run for 100 ns, with the V-rescale and Parrinello–Rahman methods used to regulate temperature and pressure, respectively (Kawata & Nagashima, 2001). To solve the Newtonian equations of motion, the leapfrog integration method was employed with a time step of 2 fs. The Particle Mesh-Ewald (PME) method, with a Fourier spacing of 0.16 nm, was utilized to handle long-range electrostatic interactions, and the LINCS method was applied to constrain all bond lengths. Trajectories were visualized, analyzed, and animated using Visual Molecular Dynamics (VMD) software version 1.9.3 and PyMOL version 2.4.1 (Carretero-González et al., 2005). Binding free energy calculations for the compound were performed using the gmx_mmpbsa tool (
Results
Drug Active Component Screening and Target Prediction
Drug components were retrieved from the Traditional Chinese Medicine Systems Pharmacology Database (TCMSP). Components with OB ≥30% and DL ≥0.18 were considered effective, whereas those without target interactions were excluded. Through this screening process, active drug components were identified, leading to the recognition of 102 targets following gene name conversion. This selection of active components lays the foundation for subsequent disease target identification and their potential therapeutic applications, as explored in the following sections (Table 1) (Ru et al., 2014).
The Effective Components of Cinnamomum Cortex (Top 10).
Disease-related Targets
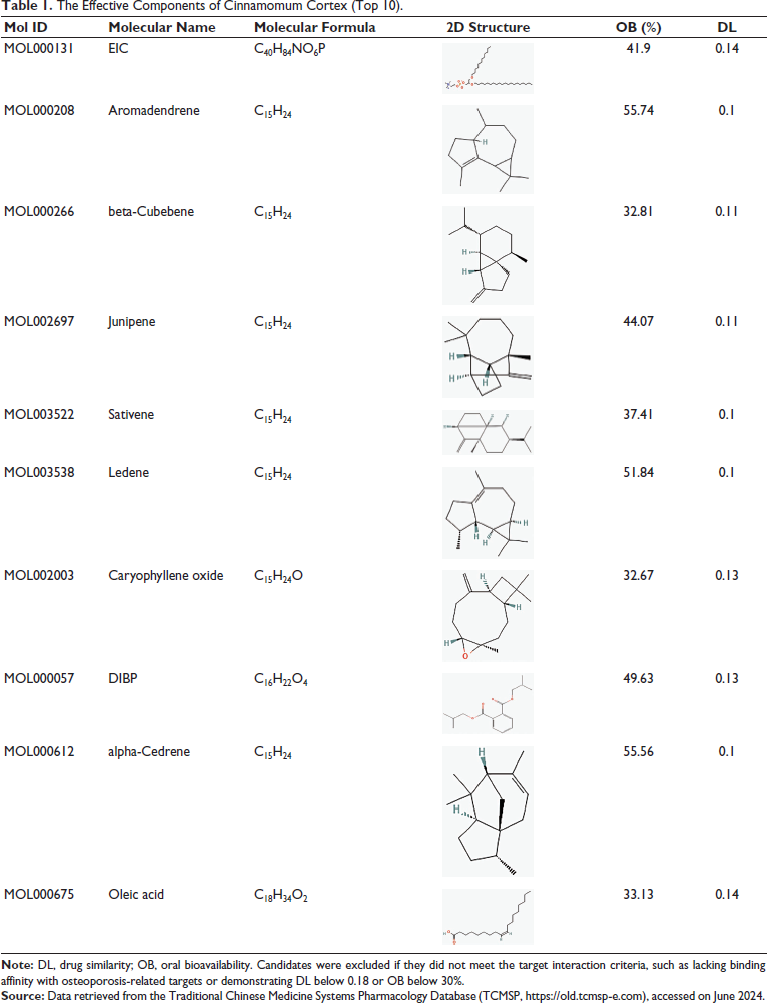
A total of 5,632 targets were retrieved from two disease-related databases: GeneCards (
Venn Diagram of Osteoporosis-related Targets and Targets of Cinnamomum Cortex.
“Drug–Target” Prediction Results
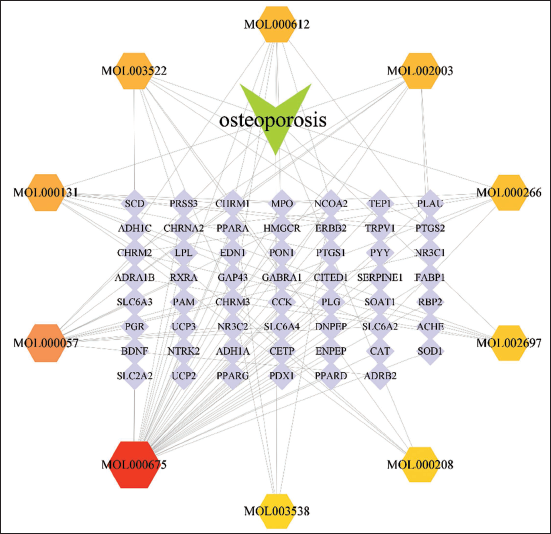
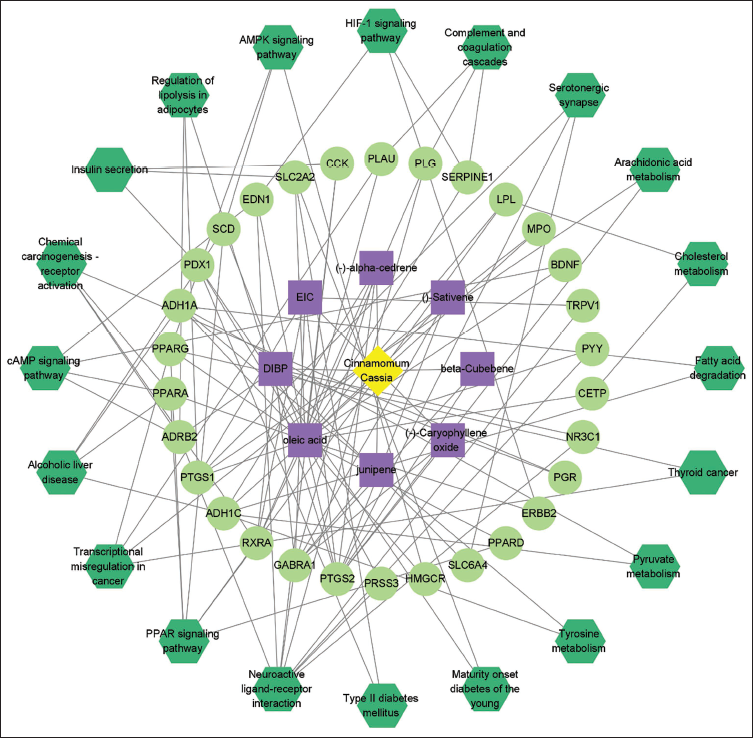
The intersection of drug-related and disease-related targets was identified, resulting in 38 overlapping drug–target genes. A “drug–target” network diagram was created using Cytoscape version 3.9.1, as shown in Figure 3. This network comprises 66 nodes and 122 edges. Key compounds were determined through the Degree algorithm available in the CytoHubba plugin. The principal compounds identified include oleic acid, DIBP, EIC, sativene, and (–)-caryophyllene oxide. Each compound is clearly linked to its respective osteoporosis-related target genes within the drug–target network. The network topology, represented by nodes and edges in Figure 3, visually demonstrates the specific binding interactions and the centrality of these compounds within the overall network. This depiction emphasizes the multi-target therapeutic role of these compounds in mediating the therapeutic effects of C. cassia on osteoporosis through a complex interaction mechanism. The recognition of these key compounds and their corresponding targets forms the foundation for constructing the PPI network, as described in the following section.
Drug–Target Network.
PPI Network of Common Targets
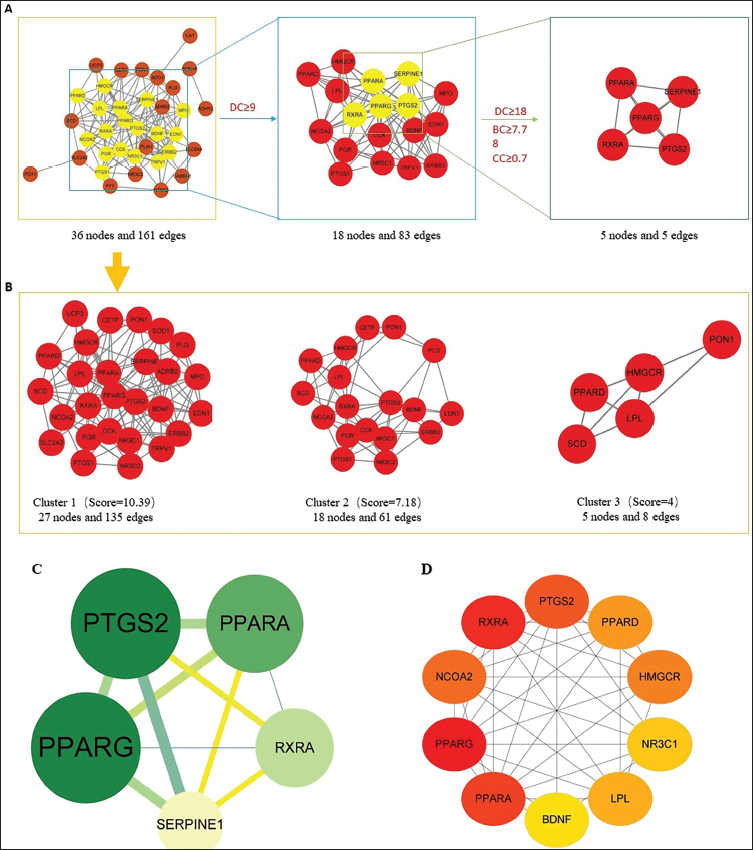
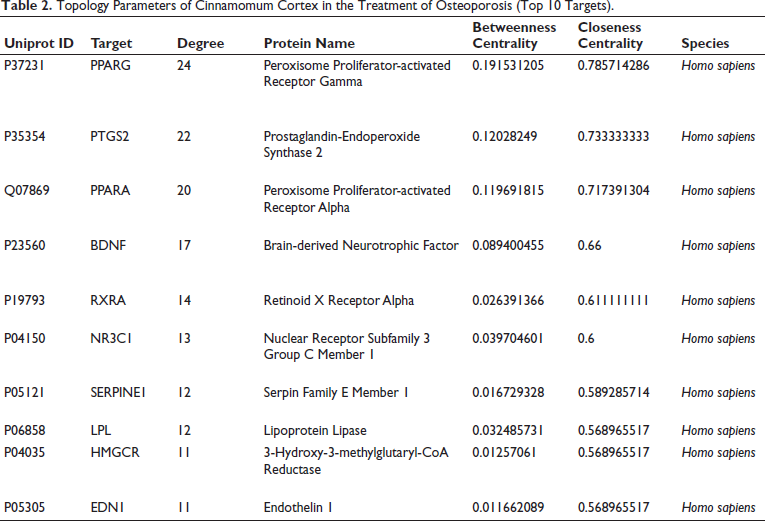
To construct the PPI network, 38 shared targets were submitted to the STRING database, resulting in a network structure comprising 38 nodes and 161 edges. Five core targets were identified based on their degree values, betweenness centrality (BC), and closeness centrality (CC), as demonstrated in Figure 4A and C. Cluster analysis, illustrated in Figure 4B, was performed using the MCODE algorithm, which yielded a highly interconnected subnetwork. The identified targets were subsequently classified into three distinct clusters. Additionally, the CytoHubba plugin was utilized to determine the top 10 hub genes by applying the Maximal Clique Centrality (MCC) algorithm, as shown in Figure 4D. The detailed topological parameters of the network are summarized in Table 2.
Elucidation of Potential Targets Through Analysis of Protein–Protein Interactions (PPIs). (A) Topological Filtering Process Within the PPI Network. (B) Analysis of the PPI Network Utilizing Cluster Analysis Through the MCODE Plugin. (C) Identification of Five Key Hub Targets from 38 Shared Targets via Degree Centrality (DC), Betweenness Centrality (BC), and Closeness Centrality (CC); the Node Size Reflects the Degree of the Target Within the Network. (D) Selection of Hub Genes from the PPI Network Employing the CytoHubba Plugin, Where Node Color Shifts from Light Yellow to Red Indicating an Increasing Degree.
Topology Parameters of Cinnamomum Cortex in the Treatment of Osteoporosis (Top 10 Targets).
Biological Function Enrichment Analysis
GO Enrichment Analysis
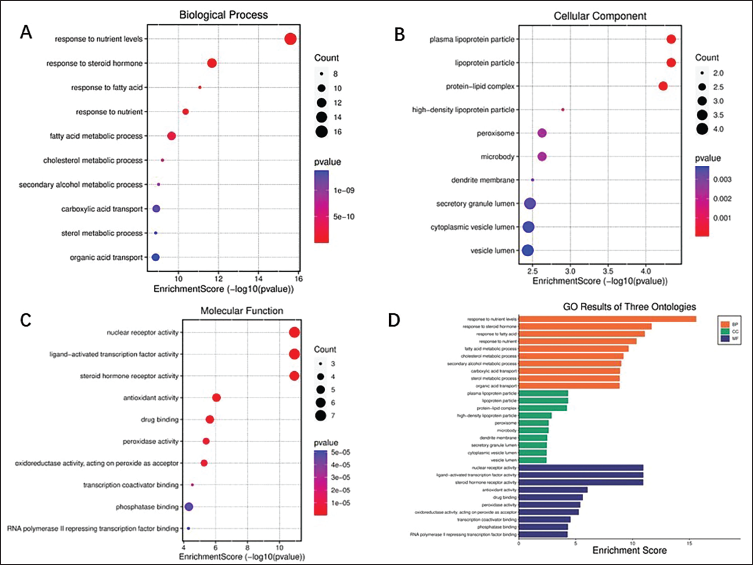
Using R software, a GO gene function enrichment analysis was conducted on the overlapping genes between drug- and disease-related targets. This analysis identified 1,795 biological process (BP) terms, 140 cellular component (CC) terms, and 231 molecular function (MF) terms. The intersecting genes derived from the active components of C. cassia and osteoporosis-associated targets were analyzed, specifically focusing on 38 shared genes between the drug and disease. The top 30 significantly enriched terms (p < 0.05) related to drug action are presented in Figure 5. In the BP category, key terms include nutrient response, steroid hormone response, fatty acid metabolism, cholesterol metabolism, secondary alcohol metabolism, carboxylic acid transport, sterol metabolism, and organic acid transport (Figure 5A). For CC, the enriched terms primarily involve plasma lipoprotein particles, protein–lipid complexes, high-density lipoprotein particles, peroxisomes, microbodies, and dendritic membranes (Figure 5B). MF terms highlight nuclear receptor activity, ligand-activated transcription factor activity, steroid hormone receptor activity, antioxidant activity, drug binding, and peroxidase activity (Figure 5C). This integrative analysis of BPs and MFs provides a foundation for elucidating the therapeutic mechanisms of C. cassia, which are further explored through the enriched signaling pathways discussed in the subsequent section.
Outcomes of Gene Ontology (GO) Enrichment Analysis. (A–C) Displayed as a Bubble Chart, the Top 10 Terms for Biological Process (BP), Cellular Component (CC), and Molecular Function (MF) in the GO Enrichment Study. (D) The Top 10 Terms for Each Category BP, CC, and MF are Depicted in a Column Chart with Colors Orange, Green, and Purple Representing Each Category, Respectively.
KEGG Pathway Enrichment Analysis
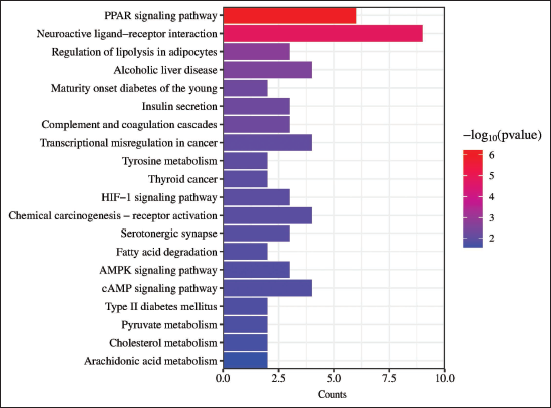
The KEGG pathway enrichment analysis was performed using R software, which identified 131 pathways related to drug treatment of diseases. The Regulation of Lipolysis in Adipocytes pathway, which includes key genes such as PTGS2, is implicated in the metabolic processes related to energy homeostasis. Osteoporosis is associated with changes in fat metabolism, and the regulation of lipolysis in adipocytes may influence bone remodeling by altering the availability of fatty acids and energy substrates required for osteoblast and osteoclast activity. Statistical significance was noted where p < 0.05, This analysis covered pathways such as the PPAR signaling pathway, HIF-1 signaling pathway, AMPK signaling pathway, and cAMP signaling pathway, as illustrated in Figure 6 and detailed in Table 3.
Bar Graph Depicting the Top 20 Pathways as Determined by Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis.
Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Results of Key Genes.
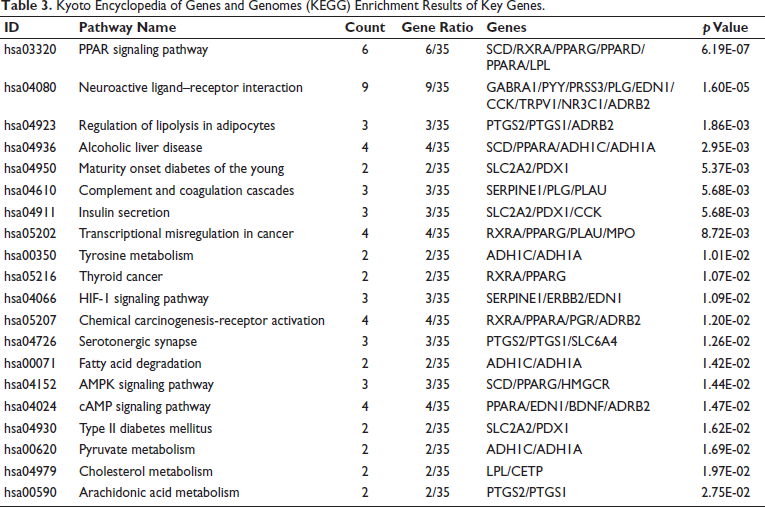
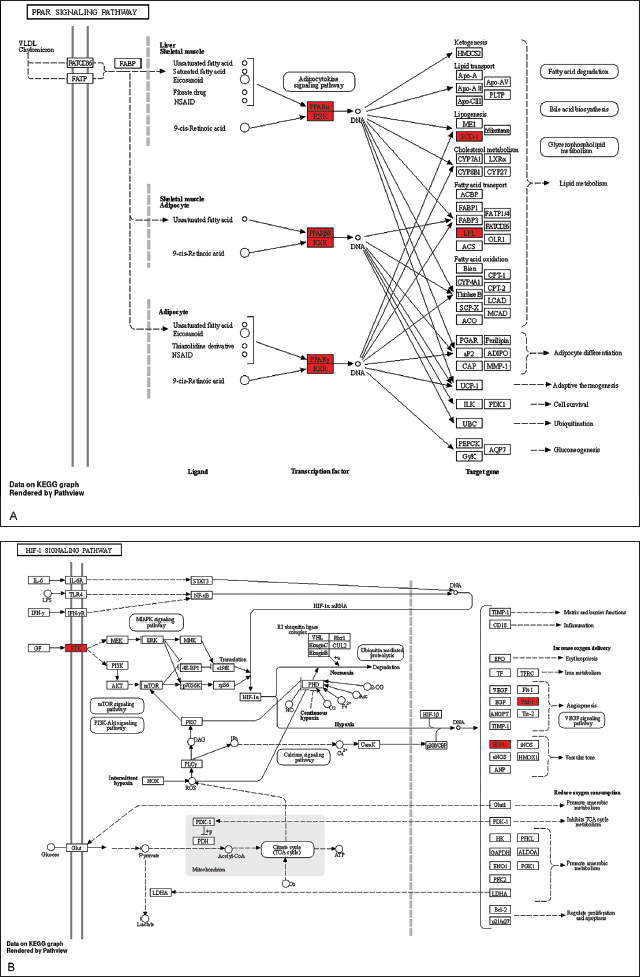
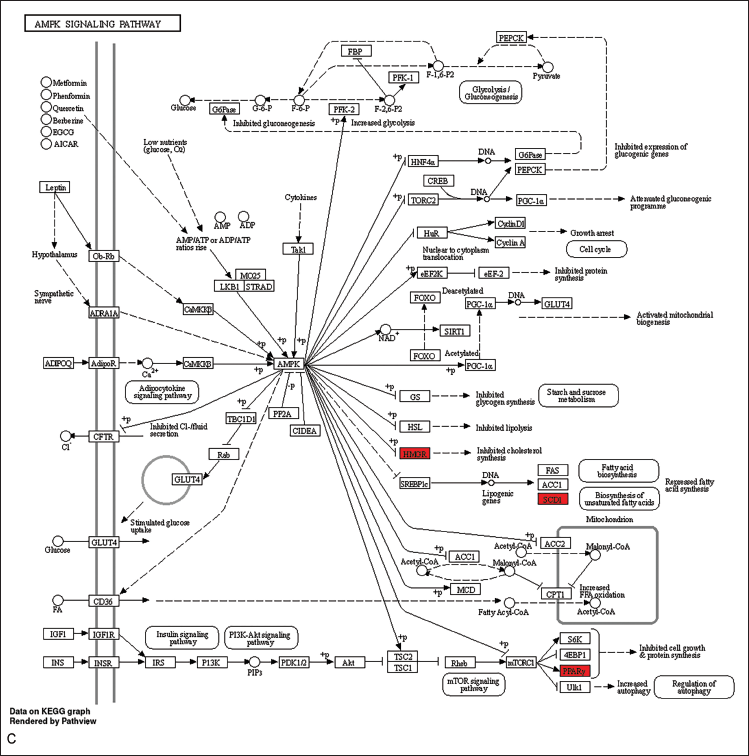
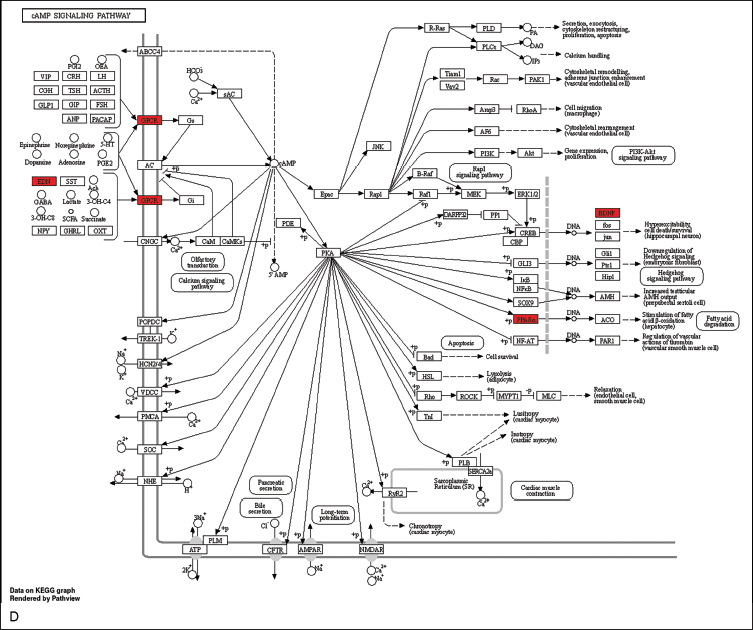
The visualization of these pathways, including the PPAR, HIF-1, AMPK, and cAMP signaling pathways, was performed to elucidate their potential therapeutic implications and was achieved using the “pathview” package in R (Figure 7A–D).
Allocation of Principal Targets Across the Most Pertinent Pathways. (A) Allocation of Principal Targets Within the PPAR Signaling Pathway. (B) Allocation of Principal Targets Within the HIF-1 Signaling Pathway. (C) Allocation of Principal Targets Within the AMPK Signaling Pathway. (D) Allocation of Principal Targets Within the cAMP Signaling Pathway. Key Targets are Represented by a Red Rectangle. The Conjectured Targets and the Genes Involved in Each Pathway are Marked in Red.
The Drug–Target–Pathway (D–T–P) Network
The top 20 pathways were selected and integrated into a Drug–Target–Pathway (D–T–P) network using Cytoscape version 3.9.1. This network, comprising 59 nodes and 119 edges (Figure 8), plays a crucial role in elucidating the mechanisms underlying of C. cassia in the treatment of osteoporosis.
Molecular Docking
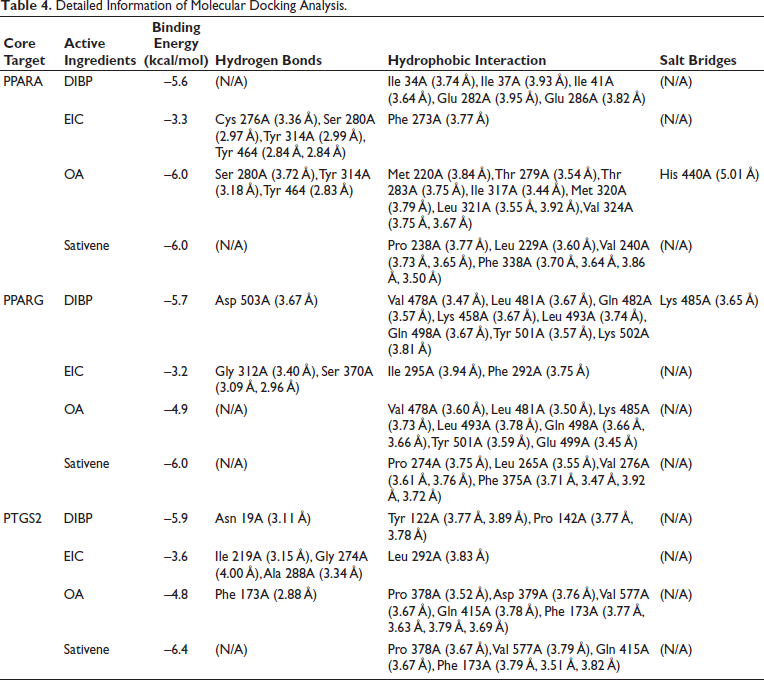
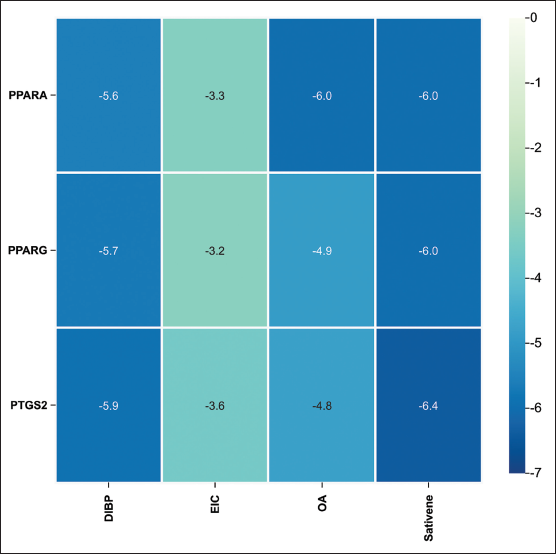
The molecular docking results provided detailed information on binding energy, interaction forces, and bond lengths, as shown in Table 4 and Figure 9. Heat maps (Figure 10) illustrated the binding energies of active compounds to the core targets, highlighting that DIBP, OA, and sativene exhibited stronger binding affinities to the three core targets. Hydrophobic interactions were identified as the primary mechanism by which the active compounds interacted with the core targets. The active pocket of PPARA consisted of amino acid residues Ile, Glu, Cys, Ser, Phe, Tyr, Met, Thr, Leu, His, Pro, and Val. The active pocket of PPARG consisted of amino acid residues Asp, Val, Leu, Gln, Lys, Tyr, Gly, Ser, Ile, Phe, Glu, and Pro. The active pocket of PTGS2 consisted of the amino acid residues Asn, Tyr, Pro, Ile, Gly, Ala, Leu, Phe, Asp, Val, and Gln. Most of the binding pockets of the targets are hydrophobic amino acid residues. Notably, the aromatic amino acid residues Phe, Trp, and Tyr in the target site were able to bind to each other with the active ingredient. This suggests that the active ingredient has some degree of influence on the fluorescence bursting effect of the target proteins (Lacativa & Farias, 2010).
Detailed Information of Molecular Docking Analysis.
Binding Energy Thermograms of Core Active Ingredients and Core Targets.
Molecular Dynamics Simulation
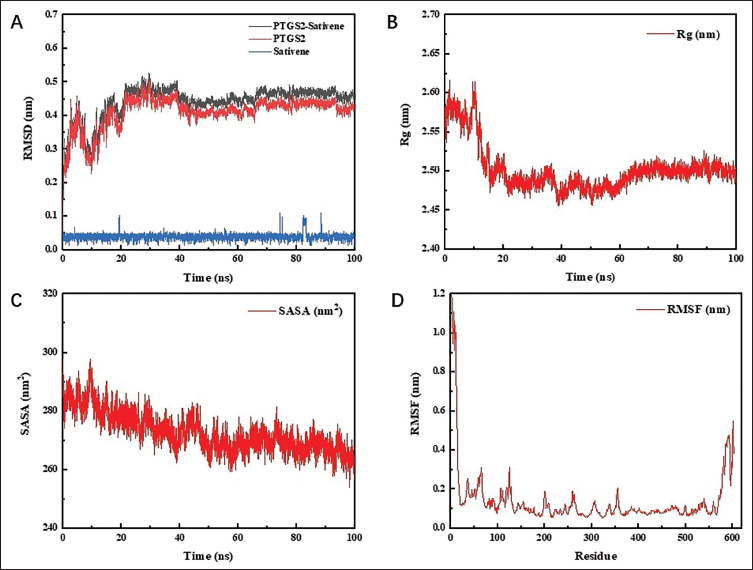
The root mean square deviation (RMSD) measures the average displacement of atomic coordinates relative to a reference model and is commonly employed to evaluate the stabilization of simulation systems. A consistent RMSD indicates equilibrium within the system, while fluctuations in RMSD reflect instability. As shown in Figure 11A, the RMSD values for PTGS2–sativene, PTGS2, and sativene were 0.440 ± 0.049 nm, 0.409 ± 0.048 nm, and 0.039 ± 0.009 nm, respectively, with minimal variation, confirming stable protein–ligand interactions. The radius of gyration (Rg), which represents the compactness of a protein during simulations and is defined as the distance between the center of mass and the outermost atoms, showed a downward trend throughout the MDS of the complex (Figure 11B). This decline suggests enhanced structural compactness. Simultaneously, the solvent accessible surface area (SASA) exhibited a similar downward trend, reinforcing the observation of increasing protein structural compactness (Figure 11C). Additionally, the root mean square fluctuation (RMSF) quantifies the extent of atomic deviations from their average positions, reflecting the time-averaged structural variability and providing insight into protein regions’ flexibility. According to Figure 11D, the average RMSF for the simulated protein complex stood at 0.139 nm, with notably stable fluctuations at the active site, indicating robust PTGS2 protein interaction with sativene.
RMSD, Rg, SASA, and RMSF of MDS. (A) The RMSD of PTGS2–Sativene. (B) The Rg of PTGS2–Sativene. (C) The SASA of PTGS2–Sativene. (D) The RMSF of PTGS2–Sativene.
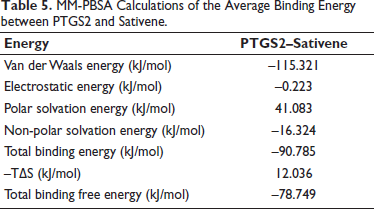
To analyze the dynamics of protein–ligand interactions, the gmx-mmpbsa script (
MM-PBSA Calculations of the Average Binding Energy between PTGS2 and Sativene.
Discussion
Osteoporosis is a systemic bone disorder characterized by reduced bone density and diminished bone quality, resulting from a range of contributing factors, that increases the susceptibility to fractures (Lane et al., 2000; Warriner et al., 2011). This network pharmacology research identified oleic acid (Sales-Campos et al., 2013), DIBP, EIC, sativene, and (–)-caryophyllene oxide as key components (Kasonga et al., 2019; Schaepe et al., 2017). Studies have shown that dietary intake of oleic acid improves bone metabolism, a crucial bone marker (Fonolla-Joya et al., 2016). This may be related to oleic acid stimulating nuclear factor κB ligand and increasing the activity of RANKL in bone cells (Fischer & Haffner-Luntzer, 2022; Song et al., 2019; Tsukasaki, 2021). Another study in rats found that oleic acid affects the formation of melatonin and triglycerides, hindering melatonin’s inhibitory effect on triglyceride accumulation, suggesting its role in osteoporosis intervention via melatonin receptor-mediated mechanisms (Sanchez-Hidalgo et al., 2007). Oleic acid influences mesenchymal stem cell lineage differentiation by modulating the content of bone marrow adipose tissue, a process potentially mediated through interactions between gut microbiota and their metabolic byproducts (Almalki & Agrawal, 2016; Park et al., 2020). Oleic acid intake markedly decreases the number of bone marrow adipocytes, promotes the formation of osteoblasts, and modulates osteoprotegerin expression at both mRNA and protein levels (Zhang et al., 2022). DIBP, a compound strongly associated with osteoporosis progression, suppresses the proliferation of GC-1 spg cells through autophagy activation and the downregulation of the PI3K-AKT and ERK signaling pathways, indicating its potential in osteoporosis regulation through multiple pathways (Kim et al., 2024). DIBP also regulates hormone levels, influencing bone growth in rats, suggesting a new regulatory direction in osteoporosis treatment (Sedha et al., 2015). Sativene, a sesquiterpene compound with a unique bicyclic octane core carbon skeleton, indicates an unusual biosynthetic pathway and promotes biological growth (Li et al., 2020). Caryophyllene oxide is strongly associated with the production and accumulation of intracellular reactive oxygen species and lipid peroxidation, phenomena frequently observed in osteoporosis among elderly cancer patients (Drake, 2013; Xiu et al., 2022). Studies indicate that caryophyllene oxide mitigates osteoporosis progression in cancer patients by modulating autophagy, increasing the levels of NCOA4 and LC3 proteins in bone cell tissues, and elevating serum Fe2+ and malondialdehyde concentrations. Additionally, it suppresses the expression of NRF2, GPX4, HO-1, and FTH1, thereby reducing the inhibitory effects of serum GSH and hydroxyl radicals. Furthermore, caryophyllene oxide enhances bone deposition within tumor tissues, contributing to the suppression of tumor growth and the delay of osteoporosis (Shabana et al., 2023; Xiu et al., 2022).
An integrated evaluation of the component-target network and the PPI network in the study suggests that PPARG, PTGS2, PPARA, BDNF, RXRA, NR3C1, SERPINE1, HMGCR, and EDN1 are potential core targets in the treatment of osteoporosis with cinnamon (Ghallab & Seddek, 2020; Jiang et al., 2020; Li, Wang et al., 2023). Evidence indicates that PPARA protein may hinder osteoporosis drug therapy, potentially due to non-expression at binding sites, while RXRA protein specifically binds to ADRB2, facilitating drug effectiveness against osteoporosis (Kim et al., 2019). Increasing evidence shows that proteins like HMGCR and EDN1, which play significant roles in regulating cell apoptosis, differentiation, and tumor suppression, are upregulated in osteoporosis patients (Ren & Zhou, 2024; Zheng et al., 2020; Zhou et al., 2021). Modulating these proteins can reduce cell apoptosis and inflammation, thus treating osteoporosis (Li, Pang et al., 2023; Muñoz et al., 2020). Osteoporosis is often associated with fluctuations in calcium ion levels at synapses. Activating PTGS2 can reduce calcium levels, enhancing tissue cell plasticity and thereby treating osteoporosis (Kelly et al., 2013).
The findings from the GO enrichment analysis indicated the roles of C. cassia in managing osteoporosis primarily involve interactions with various BPs related to nutrient levels, steroid hormones, fatty acids, and nutrition, such as fatty acid metabolic processes, cholesterol metabolic processes, secondary alcohol metabolic processes, and the transport of carboxylic and organic acids. In terms of CCs, significant associations were observed with plasma lipoprotein particles, protein–lipid complexes, high-density lipoprotein particles, peroxisomes, microbodies, and cristae membranes. MFs primarily involve nuclear receptor activity, ligand-activated transcription factor activity, steroid hormone receptor activity, antioxidant activity, drug binding, and peroxidase activity (Gennari et al., 2007; Lippi et al., 1998; Wang et al., 2023). Furthermore, KEGG enrichment analysis elucidates that C. cassia’s intervention in osteoporosis predominantly activates the PPAR, HIF-1, AMPK, and cAMP signaling pathways (Chen et al., 2017).
Research has established a strong correlation between immunity, inflammation, redox processes, ion transport, apoptosis, and the progression of osteoporosis (Iantomasi et al., 2023). Signaling pathways involved in inflammation, including the PPAR, HIF-1, and AMPK pathways, are closely linked to inflammatory responses within the autoimmune system (Bai et al., 2023; Hu et al., 2023; Tao et al., 2021). These signaling pathways immediately participate in autoimmune inflammatory responses upon activation, possibly related to the activation of relevant effective factors. Regulating these signaling pathways can modulate the “neuroimmune inflammation” process of osteoporosis (Lacativa & Farias, 2010). Also, recent research has demonstrated that C. cassia mitigates osteoporosis by inhibiting osteoclast differentiation and activity via suppression of the NF-κB and MAPK signaling pathways. Notably, it reduces the expression of pro-inflammatory cytokines, including TNF-α and IL-1β, which play critical roles in driving osteoclastogenesis. Additionally, C. cassia influences the RANKL/OPG ratio, enhancing osteoblast function and inhibiting osteoclast activity, which collectively contributes to maintaining bone homeostasis (Liu et al., 2021; Zhang et al., 2019).
Molecular docking analysis confirmed favorable binding interactions between the principal targets and active molecules. Stability assessments of PTGS2–sativene complexes, as evidenced by MDSs, indicated significant stability. This study highlights the potential role of sativene in contributing to the therapeutic effects of C. cassia in the treatment of osteoporosis.
This study advances the understanding of C. cassia in osteoporosis treatment by integrating network pharmacology, molecular docking, and dynamics simulations. Unlike prior research focusing on single components, our multi-target approach provides a comprehensive view of its therapeutic effects through anti-inflammatory and bone metabolism pathways. By combining traditional medicine insights with advanced computational tools, this study underscores the value of network pharmacology as a powerful tool in advancing traditional Chinese medicine research.
Despite uncovering some key preliminary insights, this study has certain limitations that should be acknowledged. The exploration of C. cassia’s role in osteoporosis therapy was solely conducted using sophisticated bioinformatics and computational methodologies. Consequently, the predictions’ reliability and accuracy require confirmation through additional in vivo animal testing (Wang et al., 2024).
Conclusion
This study integrates systematic pharmacology with molecular docking and MDSs to explore the detailed therapeutic mechanisms of C. cassia in the treatment and management of osteoporosis. Findings from the experiment proved that some bioactive compounds found in C. cassia are oleic acid, DIBP, EIC, sativene, (–)-caryophyllene oxide, which targets only PPARG, PTGS2, and PPARA. It thus activates a few key signaling pathways, that is, PPAR, HIF-1, AMPK, and cAMP, outlining the molecular basis of C. cassia effectiveness in the treatment of osteoporosis. This study emphasizes the multi-targeted and pathway-driven therapeutic strategies employed in traditional Chinese medicine for the treatment of chronic diseases. The study further demonstrates the holistic concept of traditional Chinese medicine and gives theoretical underpinning for future clinical research and application.
The present study findings add more light to the detailed characteristics of the pharmacodynamic properties of C. cassia and its mechanisms in the therapy of osteoporosis and provide the base for following in vivo animal research.
Footnotes
Abbreviations
DL: Drug-likeness; OB: Oral bioavailability; DC: Degree centrality; BC: Betweenness centrality; BP: Biological process; CC: Cellular component; MF: Molecular functions; MCC: Maximal Clique Centrality; PPI: Protein–protein interaction; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; MDT: Molecular docking technology; MDS: Molecular dynamics simulation.
Consent for Publication
Not applicable.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval and Informed Consent
Not applicable.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.