
Abstract
Background
The discovery of new drug targets is a critical and challenging aspect of drug development. Fascaplysin, a marine natural product, has shown potential in various biological applications, but identifying suitable protein targets for such compounds remains a complex task.
Objectives
This study aims to identify new candidate protein targets for fascaplysin by analyzing the expected targets of structural analogs, utilizing both molecular docking analysis for prediction and in vivo experiments for validation.
Materials and Methods
We utilized a public database to classify structurally similar analogs to fascaplysin into four groups based on their scaffold structures. The predicted protein targets were organized by scaffold structure and assessed for potential binding with fascaplysin through molecular docking analysis. In adult zebrafish, we employed quantitative polymerase chain reaction (qPCR) to detect expression changes in downstream signaling genes of the selected candidate targets, providing a predictive measure of interaction and biological effect.
Results:
A total of 93 common targets were identified across all groups, categorized based on protein characteristics. The molecular binding affinities between fascaplysin and 37 selected candidates, comprising 14 kinases and 23 receptors, were quantified. Molecular docking analyses highlighted MAPK8, MAP2K1, ERBB2, and JAK3 as novel drug target candidates. In adult zebrafish, fascaplysin modulated expression changes in downstream genes of these targets. This indicates the applicability of fascaplysin in treating diseases associated with these targets, revealing promising therapeutic potential.
Conclusion:
The in silico analysis method used in this study provides an efficient and cost-effective way to identify protein target candidates for drugs, using structural similarities and molecular docking to make the target validation process in drug development faster and easier. Integration of in vivo observations in adult zebrafish, specifically the modulation of downstream gene expression of candidate targets, complements in silico findings, enhancing the reliability of target identification. These results not only identify specific targets for fascaplysin but also establish a flexible framework that can be applied to other compounds, potentially expediting the drug discovery process.
Keywords
Introduction
The exploration of novel therapeutic agents remains a pivotal focus in pharmaceutical research (Danhof et al., 2018), particularly in drug target identification, which is crucial for precision medicine advancements (Dugger et al., 2018). Traditional methods in drug target discovery, characterized by intensive labor, prolonged durations, and high costs, often involve extensive experimental procedures with variable results (Harvey et al., 2015). In contrast, computational approaches, especially those leveraging structural similarities among chemicals, offer an efficient, streamlined alternative (Sliwoski et al., 2014). These methods facilitate the prediction of potential protein targets, enhancing the efficiency of drug development (Agamah et al., 2020).
Derived from the marine sponge Fascaplysinopsis Bergquist in 1988 (Manda et al., 2016), fascaplysin is a benzoyl-linked β-carboline alkaloid known for its inhibition of cyclin-dependent kinase 4 (CDK4). This action hinders the proliferation and metastasis of certain cancer cells, including small-cell lung (Hamilton, 2014) and ovarian cancers (Chen et al., 2017). Additionally, its ability to inhibit acetylcholinesterase suggests potential for Alzheimer’s disease treatment (Bharate et al., 2012). As a multitarget drug, fascaplysin can act on several biological targets simultaneously, offering advantages for treating complex diseases by potentially improving bioavailability and pharmacokinetic profiles (González et al., 2019). Identifying and confirming its specific targets are crucial for its safe and effective use in treating multi-pathway diseases.
The objective of this study is to utilize in silico analysis to discover new protein targets for fascaplysin. Focusing on the protein targets of structurally similar compounds (Bhrate et al., 2012), the study aims to identify proteins that might interact with fascaplysin. This hypothesis-driven approach suggests that compounds with structural analogies often share protein targets (Wang et al., 2013), which forms a basis for predicting the potential protein interactions of fascaplysin.
Using a public database (Daina et al., 2019; Davies et al., 2015; Kim et al., 2023; Wishart et al., 2018), we identified fascaplysin analogs and categorized them based on scaffold structures and predicted protein targets. This categorization enabled a focused analysis of the protein targets associated with each analog group, aiming to identify common proteins as potential targets for fascaplysin. Molecular docking analyses were conducted to explore the binding affinities (Meng et al., 2011) between fascaplysin and these protein candidates.
Integrating zebrafish models into drug evaluation offers a dynamic and cost-effective method to validate computational predictions in vivo. Zebrafish, due to their genetic similarity to humans and transparent embryos, allow for rapid, visually accessible assessments of drug effects and toxicity (MacRae & Peterson, 2015). This approach bridges the gap between in silico analyses and human clinical trials, providing a quicker path to understanding drug interactions and safety profiles within a biological system.
The outcomes of this study are expected to contribute to drug discovery by providing a method for target identification that reduces the need for extensive experimental validation. Additionally, this research will include in vivo validation to confirm the targets predicted through computational analyses for fascaplysin. Combining computational predictions with in vivo validation offers a methodological advancement in the field of drug target discovery, potentially improving the efficiency of this critical phase of drug development by providing a deeper understanding of drug-target interactions.
Materials and Methods
Query Molecule
In molecular biology research, we employ query molecules embedded with bioactive substances, notably fascaplysin and its analog derivatives. These query molecules are designated to interact with specific biological targets, which may be either previously characterized or not yet identified. When the biological target of a query molecule is known, it serves as a reference standard. This standard is instrumental in facilitating the prediction of potential biological targets for adjacent molecules, especially for those with currently undefined targets.
Chemical Similarity Searching
In this study, we explored the analogs of fascaplysin by querying ChEMBL (Davies et al., 2015), PubChem (Kim et al., 2023), and ZINC (Irwin et al., 2020) databases. These databases provided the common names and chemical structures of a large number of bioactive molecules, with ChEMBL and PubChem being the primary sources. Initially, we utilized these databases for structure similarity searches, identifying 28 fascaplysin analogs with a structural similarity of at least 40% in the ChEMBL database. PubChem and ZINC databases were also employed for additional chemical similarity searches.
Data Collection
We categorized fascaplysin and its 28 analogs into four types (A–D) based on their scaffold structures. Subsequently, we gathered information on targets and structure–activity relationships (SAR) from ChEMBL, PubChem, DrugBank (Wishart et al., 2018), and SwissTarget (Daina et al., 2019). DrugBank and the SwissTarget database provided data on known targets, while target predictions were derived from PubChem. We specifically selected predictive targets that demonstrated a high likelihood (70–90% confidence) of interaction with fascaplysin and its analogs, as indicated by their “active” status in the ChEMBL database.
For instance, to assess the predictive accuracy of our method, we applied the target prediction for fascaplysin from the ChEMBL database to 352 yet-to-be-identified targets. Among these, 34 targets showed direct interaction with fascaplysin. Additionally, 93 targets, which were consistently present across all four scaffold types, were identified as predictive targets for fascaplysin.
Protein and Ligand Structure Preparation
Before conducting molecular docking, we sourced the three-dimensional (3D) structure of the protein from the Protein Data Bank (Burley et al., 2021). The protein was then processed by separating the ligand, adding polar hydrogen atoms and Kollman charges, and removing water molecules. The protein was further refined using Discovery Studio Visualizer (version 21.1.0, BIOVIA, San Diego, CA, USA), which minimized the structure and saved it in PDB format for subsequent analysis (Trott & Olson, 2010).
Molecular Docking
Molecular docking was conducted using AutoDock Vina 4.2.1, interfaced with PyRx, applying the methodology established by Trott and Olson (2010). We utilized the prepared target protein structure and fascaplysin as the receptor and ligand, respectively. The docking process represented both the targets and fascaplysin within a grid space defined by a universal force field. The docking efficiency was evaluated based on the docking energy of each receptor-ligand complex. The highest-ranking conformations of the target-fascaplysin complexes were obtained in PDBQT format. Finally, Discovery Studio Visualizer was employed to identify and visualize the interaction sites between the target and the ligand.
Protein–protein Interaction (PPI) Networks
The STRING database (Szklarczyk et al., 2017) was used to predict protein interactions and drug-target relationships, with molecules as nodes and interactions as edges, to uncover molecular mechanisms, analyzed with STRING version 11.5.
Enrichment Analysis
Initially, the candidate targets were uploaded to DAVID Bioinformatics Resources (DAVID database,
Zebrafish Husbandry and Drug Treatment
The maintenance and experimental procedures involving zebrafish were conducted in accordance with the guidelines and regulations of the Kyung Hee University Committee on Animal Research with approval (Protocol number #KHSASP-21-302). Experiments were conducted by the method of previous studies (Park & Kim, 2020). Briefly, 12 male zebrafish were divided into the control group, ethanol group, and fascaplysin group. The ethanol group was exposed to 0.5% ethanol (v/v), and the fascaplysin group was exposed to 0.5% ethanol with 100 nM of fascaplysin (Merck, Cat.341251, Rahway, NJ, USA) for 2 days.
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
Ribonucleic acid (RNA) was isolated from zebrafish liver using Trizol® (Invitrogen, Cat. 15596-026) and reverse-transcribed with SuperScript™ III (Invitrogen, Cat. 18080-044). RNA from control and ethanol-treated zebrafish, with or without fascaplysin (n = 3/group), was pooled for complementary deoxyribonucleic acid (cDNA). RT-qPCR used 50 ng cDNA and gene-specific primers (Table 1) for 45 cycles. GAPDH served as the reference gene, with relative messenger RNA (mRNA) changes calculated via the double delta Ct method (Park & Kim, 2020), in triplicates for each gene.
Primer Sequences used for Quantitative Polymerase Chain Reaction (qPCRs).

Statistical Analysis
Statistical analyses used GraphPad Prism 9 (GraphPad Software Inc., CA, USA), employing Student’s t-test for two-group comparisons and Tukey’s test for multiple groups, with significance at p < 0.05. Data are presented as mean ± standard deviation, with a minimum sample size of n = 3.
Results
Scheme of Novel Drug-target Interactions for Fascaplysin using Structural Analogs and Bioinformatic Databases
Figure 1 describes the research methodology for identifying novel drug-target interactions for fascaplysin utilizing web-based bioinformatics databases. This process involves analyzing chemical substances and biological activities, beginning with the extraction of targeting information. A key step is the identification of compounds structurally similar to fascaplysin, specifically those with a similarity score of 40% or higher. This criterion is crucial for selecting analogs likely to exhibit structurally similar activities.
Predicting New Drug-target Interactions for Fascaplysin through Chemoinformatic Analysis. This Diagram Outlines the Approach for Identifying New Targets for Fascaplysin, Involving the Search of Major Databases (ChEMBL, PubChem, DrugBank, and SwissTarget) for Compounds Structurally Similar to Fascaplysin (Similarity Threshold: 40%). The Targets of these Analogs are then Examined to Predict Potential New Interactions for Fascaplysin.
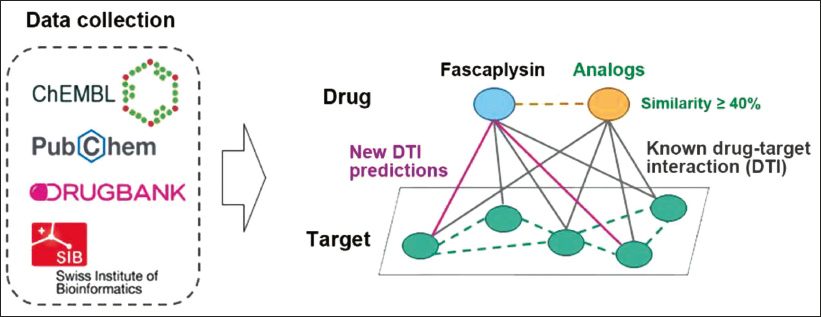
Identifying analogs leads to the analysis of predicted targets, which is crucial for understanding potential biological pathways and molecular interactions involving fascaplysin. The aim is to discover novel drug-target interactions under the assumption that structural similarity suggests similar biological activities. This approach enhances knowledge of molecular interactions with fascaplysin and investigates new therapeutic applications.
Classification of Fascaplysin and its Analog Structures
The structure of fascaplysin includes a polycyclic aromatic scaffold with interconnected rings, contributing to its stability and reactivity (Figure 2). The presence of a carbonyl group plays a significant role in defining its chemical attributes. Together, these structural components are essential in influencing the chemical and biological functionality of fascaplysin, with the scaffold enhancing stability and the carbonyl group enabling various chemical interactions.
Structural Classification of Fascaplysin Analogs. The Figure shows the Molecular Structure of Fascaplysin and its Analogs, Categorized into Types A–D Based on their Scaffold Structure (Detailed Information about the Chemical Structure is Available in Supplementary Tables 1–4).
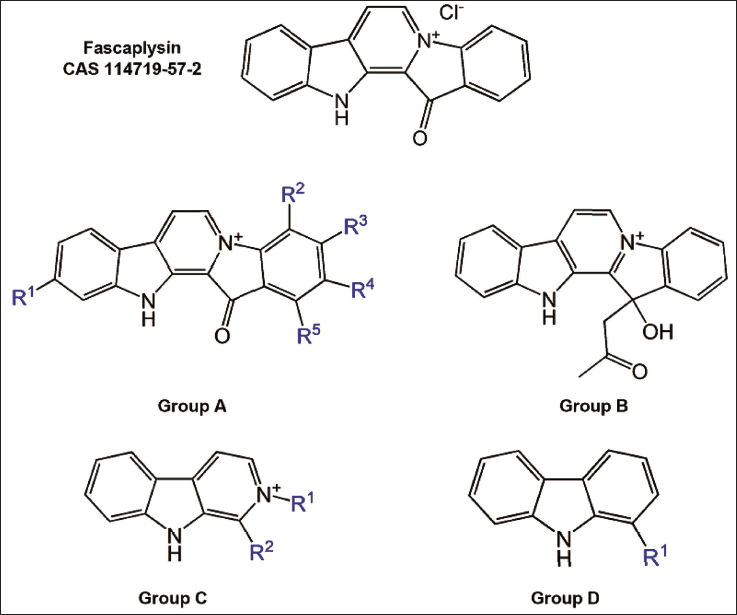
In this study, we classified 28 fascaplysin analogs into four distinct categories according to their polycyclic structure and functional groups (Figure 2 and Supplementary Table 1). These analogs are unified by foundational polycyclic aromatic structures, incorporating nitrogenous heterocycles. Type A (SMILES: O=C4C3=C2NC1=CC([R1])=CC=C1C2=CC=[N+]3C5=C([R2])C([R3])=C([R4])C([R5])=C45) analogs are characterized by an aromatic system with a nitrogenous cation and multiple substituents (R1–R5), inclusive of a proximal carbonyl group (Supplementary Table 2). Type B (SMILES:OC(C5=C4C=CC=C5)(CC(C)=O)C1=[N+]4C=CC3=C1NC2=CC=CC=C23), while retaining the fundamental polycyclic and nitrogen-rich framework, integrates a carbonyl group within an ester functional group, thereby introducing an additional ring system (Supplementary Table 2). Type C (SMILES: [R2]C3=C2NC1=CC=CC=C1C2=CC=[N+]3[R1]) diverges by excluding the carbonyl functionality and comprises two substituents (R1, R2), resulting in distinct chemical properties (Supplementary Table 3). Type D (SMILES: [R1]C3=C2NC1=CC=CC=C1C2=CC=C3) is marked by the absence of both the nitrogenous cation and the carbonyl functionality but includes a singular substituent (R1), which imparts unique modifications to physicochemical properties (Supplementary Table 4). This classification highlights the structural heterogeneity of fascaplysin analogs and their significant impact on modulating chemical reactivity and biological potency.
Comparison of Pharmacological Characteristics of Fascaplysin and its Analogs
In this study, we conducted a comprehensive comparative analysis of the chemical properties of fascaplysin and its 28 analogs. Our focus was on six key parameters crucial for drug efficacy: Molecular weight (Figure 3A), ALogP (Figure 3B), LogD at pH 7.4 (Figure 3C), the number of hydrogen bond donors (Figure 3D), acceptors (Figure 3E), and the polar surface area (Figure 3F). These properties play a crucial role in defining the interaction dynamics between compounds and biological targets.
Comparative Analysis of Chemical Properties of Fascaplysin and its Analogs. This Figure Comprehensively compares the Chemical Properties of Fascaplysin and its Analog, Which are Classified into Types A–D Based on their Scaffold Structures. Each Type is Represented with a Unique Color Scheme for Clarity. The Comparison includes (A) Molecular Weight Distribution, (B) ALogP Values, (C) LogD Values at pH 7.4, (D) the Number of Hydrogen Bond Acceptors, (E) the Number of Hydrogen Bond Donors, and (F) Polar Surface Area. All Data are Aligned Based on their Similarity to Fascaplysin.
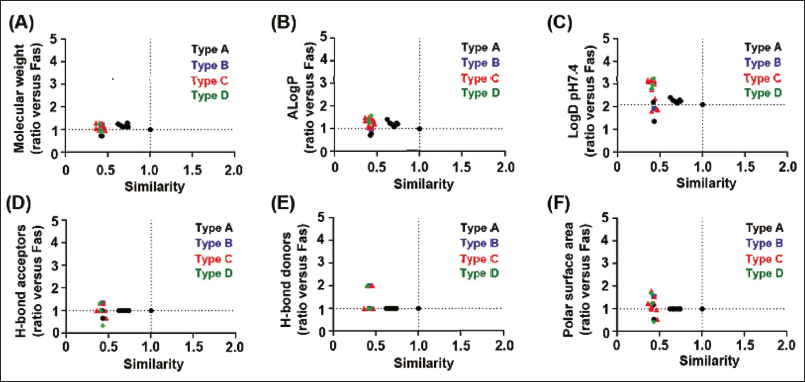
Our result revealed significant chemical similarities between fascaplysin and its analogs. Structural analysis, based on the molecular structure of fascaplysin, consistently showed comparable properties across these compounds. This similarity suggests a uniform profile in lipophilicity, solubility, and molecular interaction potential, which are key factors in drug-target binding dynamics. Therefore, we suggest that fascaplysin and its analogs are likely to exhibit similar interactions with biological targets.
Comparison of Fascaplysin and its Analogs with Predictive Targets
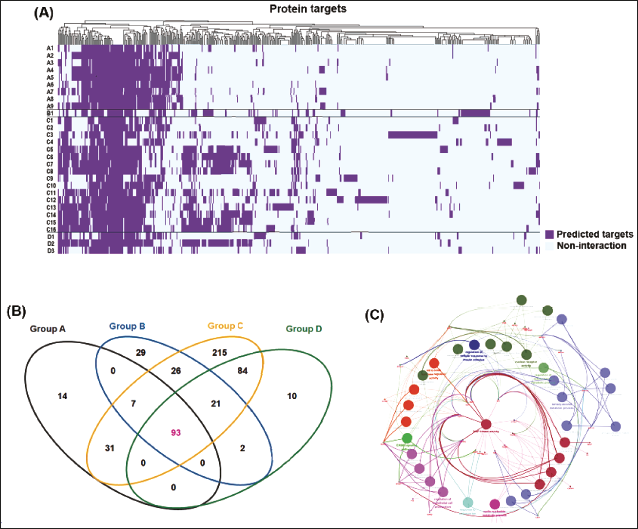
The analysis began by creating a heatmap to showcase the target profiles for fascaplysin and 28 analogs, positioning fascaplysin prominently, followed by its structurally diverse analogs. Columns in the heatmap displayed a range of predicted targets from in silico methods, with color intensity indicating the strength of predicted interaction (Figure 4A, Supplementary Table 5 provides the target list). This visualization helped identify patterns in target affinity within the fascaplysin family, revealing both unique and common pharmacological interactions. A Venn diagram was then used to show predictive target overlaps for scaffold groups A–D, uncovering 93 targets predicted in common (Figure 4B), indicating a potentially conserved action mechanism tied to the core structures of fascaplysin and its analogs. Functional analysis of these 93 targets, using the ClueGO tool (Bindea et al., 2009), pinpointed key biological processes and pathways, notably highlighting mitogen-activated protein kinase (MAPK) activity as a significant shared target feature (Figure 4C), suggesting the MAPK signaling pathway as a primary biological effect mediator for fascaplysin and its analogs.
Analysis of Predicted Targets for Fascaplysin and its Analogs. (A) The Heatmap Displays the Predicted Target Profiles, with Protein Targets on the x-axis and Fascaplysin and its Analogs on the y-axis, Providing a Visual Representation of Target Affinity across the Compounds. (B) The Analogs are Classified A–D Based on their Scaffold Structures, and a Venn Diagram Illustrates the Predicted Targets for Each Group, Highlighting 93 Targets Common to all Four Groups. (C) ClueGO Analysis was Conducted to Analyze the Interrelations of Terms and Functional Groups in the Biological Networks of these 93 Overlapping Targets.
Analysis of the Binding Affinity between Fascaplysin and Candidate Targets using Molecular Docking
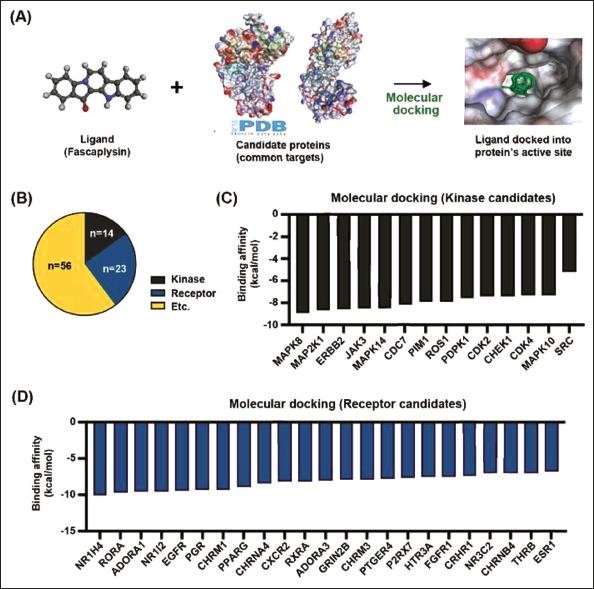
Our primary objective is to identify novel binding partners of fascaplysin, thereby enhancing our comprehension of its molecular mechanisms and potential therapeutic applications. The candidate protein structure was identified in the Protein Data Bank for the calculation of binding affinity with fascaplysin using molecular docking analysis (Figure 5A). We categorized the 93 shared targets based on their protein functions (Figure 5B). Notably, kinases and receptors were selected for molecular docking analysis due to their critical roles in cell signaling pathways and regulation. The binding affinity score offers quantitative insights into the potential of fascaplysin for interaction with novel candidate proteins. Figures 5C and D present the binding affinity of kinase and receptor candidates with fascaplysin, ranked in descending order of affinity. Among kinase candidates, MAPK8, MAP2K1, erb-b2 receptor tyrosine kinase 2 (ERBB2), and Janus kinase 3 (JAK3) demonstrated high binding affinities. In the receptor category, nuclear receptor subfamily 1 group H member 4 (NR1H4), RAR-related orphan receptor A (RORA), and adenosine A1 receptor (ADORA1) exhibited notable binding affinities with fascaplysin.
Molecular Docking Analysis of Fascaplysin with Candidate Protein Targets. (A) A Schematic Diagram Outlines the Molecular Docking Method used. Fascaplysin, Serving as the Ligand, was Docked with Candidate Protein Structures Sourced from the Protein Data Bank. (B) The 93 Targets Identified were Classified According to their Protein Functions, Revealing 14 Kinases and 23 Receptors. (C) This Panel shows the Molecular Binding Affinity between Fascaplysin and Each of the 14 Kinase Candidates. (D) The Molecular Binding Strength between Fascaplysin and the 23 Receptor Candidates.
Comprehensive Analysis of Kinase and Receptor Candidates to Fascaplysin
The PPI network for candidate receptor and kinase targets of fascaplysin was generated using the STRING and is shown in Figure 6A. This analysis incorporated 14 kinases and 23 receptors of Homo sapiens. The network was constructed with a confidence level greater than 0.4 and an enrichment p value of less than 0.01.
Identification of Potential New Targets of Fascaplysin among Kinase/Receptor Candidates. (A) Protein–protein Interaction (PPI) Analysis of these Candidates was Conducted using the STRING Database. (B) Gene Ontology (GO) Analysis was Performed on a List of 43 Kinases and Receptors, Highlighting their Biological Processes. (C) Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analysis was also Conducted for these 43 Proteins, Revealing their Involvement in Specific Biological Pathways.
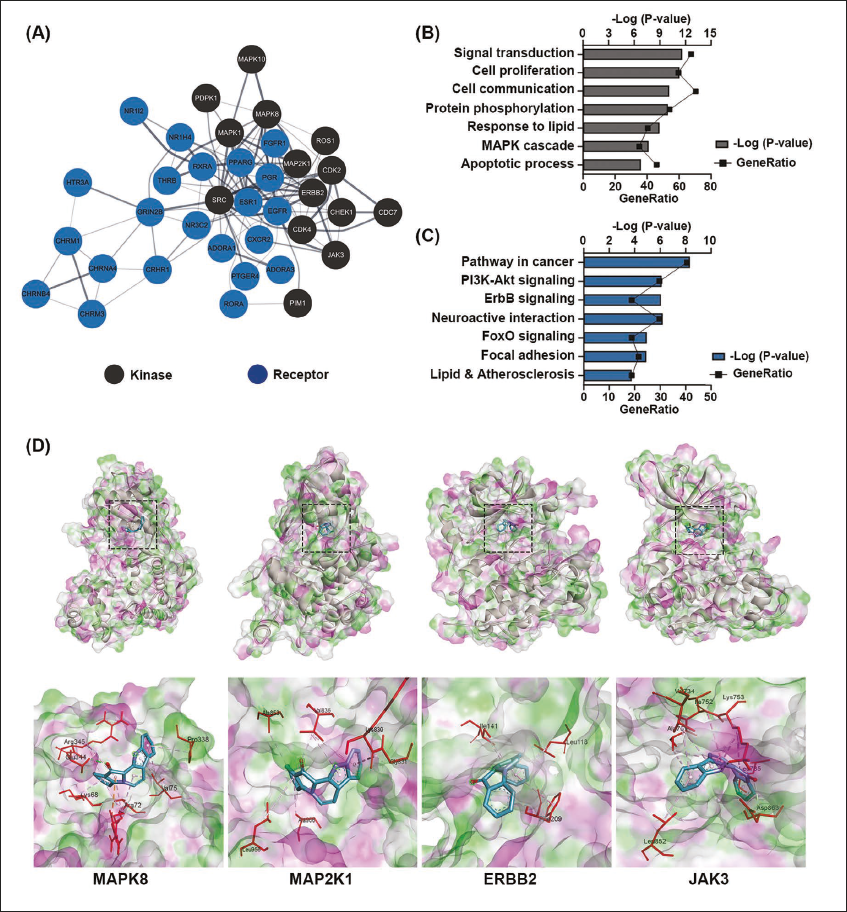
Next, GO analysis is conducted using combined kinase and receptor targets (Figure 6B). This analysis highlights significant terms related to signal transduction, cell proliferation, phosphorylation, and the MAPK cascade. The enrichment of these terms emphasizes the functional aspects of these targets and their roles in cellular processes, potentially modulated by interaction with fascaplysin. Furthermore, KEGG pathway analysis using the same set of targets (Figure 6C) reveals key pathways such as PI3K-AKT, ErbB, and cancer-related pathways. The involvement of these pathways indicates a broad spectrum of biological processes that might be influenced by fascaplysin. Figure 6D shows the 3D structure of the ligand-receptor complex formed between fascaplysin and candidate targets MAPK8, MAP2K1, ERBB2, and JAK3. These structures, obtained through molecular docking and structural modeling, offer evidence for understanding the molecular basis of interaction.
Effects of Fascaplysin on Gene Expression in Ethanol-exposed Zebrafish Liver
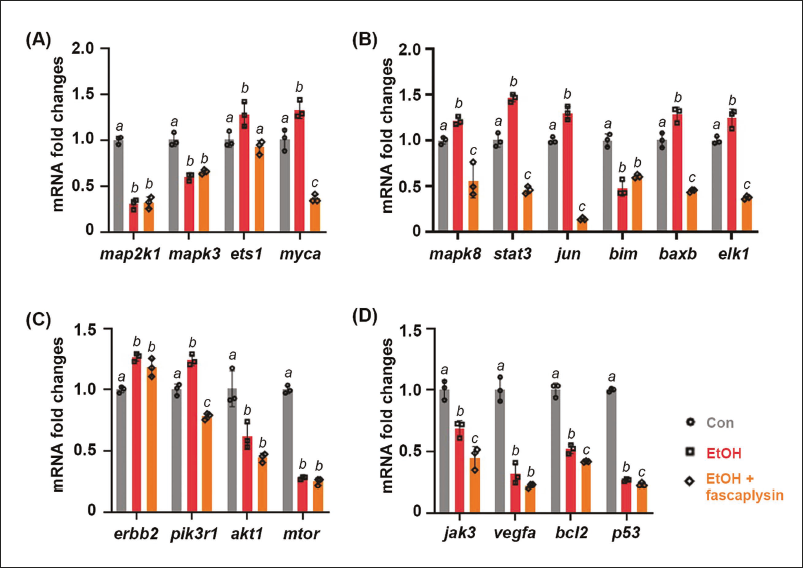
Utilizing an adult zebrafish model exposed to acute ethanol, this study validates the candidate targets of fascaplysin, initially identified through in silico approaches. Results revealed distinct gene expression patterns in response to fascaplysin. Specifically, gene expression for map2k1 and mapk3 showed no significant change compared to ethanol exposure alone, whereas genes like ets1 and myca showed notable reductions under fascaplysin treatment (Figure 7A). For genes associated with the MAPK8 pathway, such as stat3 and jun, fascaplysin treatment led to a decrease in expression, indicating potential inhibitory effects (Figure 7B). In the case of ERBB2 signaling, only pik3r1 expression decreased following fascaplysin treatment (Figure 7C), suggesting a targeted effect on this pathway. Additionally, genes related to the JAK3 pathway, upregulated by ethanol, were further suppressed by fascaplysin (Figure 7D), highlighting its strong inhibitory potential.
Fascaplysin Modulates Gene Expression in the Liver of Adult Zebrafish. Relative Messenger Ribonucleic Acid (mRNA) Expression of Downstream-related Genes in the (A) MAP2K1, (B) MAPK8, (C) ERBB2, and (D) JAK3 Pathway was Measured in the Liver Samples (n = 3) from Control and Ethanol-exposed Zebrafish with or without Fascaplysin (Final Concentration of 100 nM). The Data are Presented as the Mean ± Standard Deviation (SD); a,b,c Mean Values with Different Superscript Letters are Significantly Different among the Groups (p < 0.05).
Discussion
Fascaplysin, a kinase inhibitor with a specific affinity for CDK4, is known for its antitumor (Chen et al., 2017; Hamilton, 2014) and antibacterial (Wang et al., 2022) properties due to its interaction with the CDK4/CCND1 complex. Beyond these capabilities, fascaplysin has shown efficacy in various other conditions, such as providing analgesic effects (Johnson et al., 2017) and potential benefits in anti-Alzheimer’s disease activities (Bharate et al., 2012), thus highlighting its role as a multitarget therapeutic agent. The concept of multitarget drug development, where a single drug is designed to interact with multiple biological targets, is gaining traction in pharmaceutical research (Ramsay et al., 2018). This strategy is particularly beneficial for complex diseases such as cancer, cardiovascular diseases, and neurodegenerative disorders, which involve a myriad of biological pathways. Recent studies have explored the potential of fascaplysin in targeting other biological markers, opening avenues for novel therapeutic applications (Bhrate et al., 2012; Johnson et al., 2017; Wang et al., 2022). Its multitarget effects include modulation of other kinases, interaction with various cellular pathways, and potential influence on cellular mechanisms beyond the CDK4/CCND1 complex. Multitarget drugs like sorafenib and sunitinib demonstrate effectiveness in cancer treatment (Kim et al., 2009) by targeting multiple tyrosine kinases, inhibiting tumor growth and angiogenesis. Despite the comprehensive therapeutic potential, multitarget drugs introduce complexity in biological behavior, challenging prediction, and validation efforts (Sun et al., 2022). Research into the pharmacological profile of fascaplysin reflects the dynamic nature of drug development, emphasizing the goal of utilizing multitarget capabilities for diverse disease treatments. Exploration into complex drug actions not only advances understanding of fascaplysin but also supports the development of future multitarget therapies (Makhoba et al., 2020), offering insights into broad therapeutic implications.
In this study, we classified compounds structurally similar to fascaplysin using scaffold structures. This method is based on the principles of cheminformatics and molecular similarity (Bajorath, 2002). Our classification approach enabled a systematic method to identify potential drug targets, utilizing the concept that structural similarity often leads to similar pharmacological profiles (Wassenaar et al., 2021). Additionally, the clear chemical similarity between fascaplysin and its 28 analogs (types A–D) emphasizes the potential of structural similarity as a tool for identifying drug targets. These similarities support the idea of shared target proteins and highlight the importance of molecular structures in therapeutic effectiveness (Cheng et al., 2010). The classification of fascaplysin analogs, based on their structural and chemical properties, offers a new approach to predicting potential target proteins. By extending the known interaction profile of fascaplysin to its analogs, we can suggest new therapeutic targets and expand the scope of drug repurposing strategies. This advancement in compound classification and analysis is an important step in the field of drug discovery and development.
Prominent public databases, including ChEMBL (Davies et al., 2015), PubChem (Kim et al., 2023), ZINC (Irwin et al., 2020), and SwissTarget (Daina et al., 2019), facilitate the prediction of potential biological targets for specific compounds, thereby advancing drug development and research. This approach aligns with the growing recognition of data mining’s significance in drug discovery, as evidenced in recent publications (Bellis et al., 2011). Our investigation was underpinned by the hypothesis that compounds with analogous chemical features are likely to share common targets, a concept deeply rooted in the principle of SAR. This foundational principle in medicinal chemistry posits that structurally similar molecules frequently exhibit similar biological activities (Martin et al., 2002). In addition to these methods, we integrated an analysis of the accuracy of drug prediction targets listed in these databases. Our results were enlightening, revealing that a set of 93 targets was commonly shared by 28 analogs (Figure 4B). This finding was discerned through comprehensive heatmap and Venn diagram analysis, tools that provided visual and statistical clarity to our data. The discovery of these shared targets not only corroborates the SAR hypothesis but also demonstrates the efficacy of our data mining strategy. This approach, combining database exploration with accuracy analysis and visual data representation, offers a robust framework for identifying potential drug targets. It underscores the potential of integrating traditional medicinal chemistry principles with modern data analysis techniques to enhance drug discovery and development processes.
Molecular docking, while a commonly employed technique in the realm of drug discovery (Trott & Olson, 2010), serves as a critical step in our innovative approach to identifying potential targets for fascaplysin. Recognizing the ubiquity of molecular docking in the field, our research distinguishes itself by integrating the use of analog structure analysis for the preliminary selection of candidate targets. This methodological innovation allows us to leverage structural similarities and differences within a broader range of potential targets, refining our focus before applying molecular docking techniques for further validation. Upon establishing a narrowed field of candidates through analog structure analysis, we utilized molecular docking not as an initial screening tool but to validate and elaborate on the interactions between fascaplysin and its prospective targets. This dual-step process is particularly significant for its contribution to the repurposing efforts, as it combines the predictive power of analog analysis with the specificity of molecular docking to identify potential off-target effects and toxicity early in the development process (Ma & Zou, 2021).
Receptors and kinases, critical in biological pathways and disease mechanisms, are common drug targets. G-protein coupled receptors (GPCRs) play essential roles in cell communication and are involved in various diseases (Yang et al., 2021), making them significant targets. Kinases regulate cell functions, but dysregulation can lead to cancer (Paul & Mukhopadhyay, 2004) or autoimmune diseases (Zarrin et al., 2021). Their well-defined binding sites and pivotal role in disease pathways make these molecules attractive for drug development.
Our findings, as illustrated in Figure 6, underscore the efficacy of this approach by highlighting MAPK8, MAP2K1, ERBB2, and JAK3 as promising targets for fascaplysin. The GO and KEGG analyses further validate these targets by elucidating their roles in critical signaling pathways and cellular functions, often dysregulated in cancer (Figures 6B and D). Specifically, the identification of MAPK8 and MAP2K1 suggests the potential of fascaplysin to modulate pathways involved in cell proliferation and apoptosis, offering a novel therapeutic strategy for cancers characterized by the aberrant activation of these pathways (Guo et al., 2020; Yue & López, 2020). Furthermore, the potential targeting of ERBB2 (HER2) by fascaplysin opens new avenues for treatment in HER2-positive breast and gastric cancers, proposing a complementary or alternative mechanism to existing therapies (Gravalos & Jimeno, 2008; Yu & Hung, 2000). Similarly, the implication of JAK3 as a target extends the therapeutic possibilities of fascaplysin to include immune-related disorders, where modulation of the JAK-STAT pathway could yield significant benefits (Cornejo et al., 2009; Elwood et al., 2017).
The investigation successfully utilized a novel platform that integrates in silico predictions with in vivo validation techniques to identify therapeutic targets for fascaplysin, including MAP2K1, MAPK8, ERBB2, and JAK3. The adult zebrafish model, highly responsive to acute ethanol exposure, functioned as an effective system to observe changes in downstream gene expression (Park & Kim, 2020). qPCR analysis revealed distinctive gene expression patterns (Figure 7), indicating the selective regulatory impact of fascaplysin on specific downstream targets rather than on the initial kinase candidates. The findings offer a comprehensive view of the modulatory effects of fascaplysin, supporting its potential to treat diseases linked to these targets. While the study highlights the impact of fascaplysin on gene expression related to targeted pathways, obtaining detailed mechanistic insights necessitates further research. Additional investigations across diverse disease models are essential for a comprehensive understanding of its therapeutic scope and safety.
Conclusion
In conclusion, the study validates the use of a dual in silico and in vivo approach for the identification and verification of drug targets. The significant modulatory effects of fascaplysin on gene expression related to MAP2K1, MAPK8, ERBB2, and JAK3 not only confirm the predicted targets but also highlight its potential therapeutic applications. These insights provide a foundation for further exploration into the mechanisms of action of fascaplysin and its potential integration into treatment regimens for complex diseases.
Footnotes
Abbreviations
CCND1: Cyclin D1; CDK: Cyclin-dependent kinase; DTI: Drug-target interaction; ERBB: Erythroblastic leukemia viral oncogene homolog; ERK: Extracellular signal-regulated kinase; GO: Gene ontology; JAK: Janus kinase; JNK: c-Jun N-terminal kinases; KEGG: Kyoto encyclopedia of genes and genomes; MAPK: Mitogen-activated protein kinase; PPI: Protein–protein interaction.
Acknowledgments
The authors are thankful to Mr. Haytham Mohamedelfatih Mohamed Makki for his assistance with the maintenance and management of the zebrafish used in our study.
Data Availability Statement
The data used to support the findings of this study are included within the article and can be obtained from the corresponding author upon request.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval and Informed Consent
The maintenance and experimental procedures involving zebrafish were conducted in accordance with the guidelines and regulations of the Kyung Hee University Committee on Animal Research with approval (Protocol number #KHSASP-21-302).
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning, No. 2022R1I1A1A01053751 (to K. H. P.), No. 2021R1A2C1004133 (to N. Y. J.) and No. 2023R1A2C1003763 (to J. J.).
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.