
Abstract
Objective
Tuberculosis (TB) is among the major causes of mortality due to a single infectious bacterium. The burden of TB is higher due to multidrug-resistant Mycobacterium tuberculosis strains, which lead to treatment failures. The present study conducted in silico studies of bioactive compounds isolated from Kigelia africana (Lam.) Benth stem bark.
Methods
Pure compounds were isolated from dichloromethane/methanol stem bark extract of Kigelia africana after repeated column chromatography. The chemical structures of the isolated compounds were established based on 1H NMR, 13C NMR and 2D NMR (COSY, HSQC and HMBC) spectroscopy. In silico analyses were performed to assess the drug-likeness, pharmacokinetics and antibacterial potential of the compounds against target proteins (4QIJ and 5HKF, 5L3J and 8GZY, and 2QIL) from M. tuberculosis, Escherichia coli, and Staphylococcus aureus, respectively.
Results
Four compounds: demethylkigelin, tyrosyl butyrate, stearic acid and stigmasterol were characterized. In molecular docking studies, the compounds showed binding affinities ranging from −4.4 to −9.3 kcal/mol against target proteins 4QIJ, 5HKF, 5L3J, 8GZY and 2QIL. Stigmasterol (L4) had the highest affinity against the highest binding affinity, with a score of −9.3 kcal/mol against S. aureus protein 2QIL. It also showed strong affinities against M. tuberculosis (4QIJ and 5HKF) and E. coli (5L3J and 8GZY) targets. In silico toxicity profiling predicted tyrosyl butyrate and stearic acid to be relatively safe whereas demethylkigelin and stigmasterol showed potential respiratory and cardiotoxic effects that needs further safety evaluation.
Conclusion
Kigelia africana stem bark possesses bioactive compounds that are potential inhibitors of M. tuberculosis with good to better binding affinities and stable interactions. Future studies should validate the in vitro and in vivo bioactivity as well as toxicity of the compounds.
Introduction
Tuberculosis (TB), whose primary causative agent is Mycobacterium tuberculosis (M. tuberculosis), is among the major causes of mortality due to a single infectious bacterium. 1 Initially superseded by COVID-19, TB has now returned to be the major cause of mortality in HIV-infected patients and those related to antimicrobial resistance. 2 According to the World Health Organization, the high global burden of TB is due to multidrug (isoniazid/rifampicin)-resistant M. tuberculosis strains, leading to treatment failures when the current regimens are used.2–4 Hepatotoxicity caused by first-line antitubercular drugs used in the treatment of latent as well as active M. tuberculosis infections has also been reported. 5 Further, individuals co-infected with TB and HIV require concurrent administration of antiretroviral therapy and antitubercular drugs to improve survival. However, this combination is currently associated with challenges such as drug interactions, cumulative toxicities, and the development of TB-associated immune reconstitution inflammatory syndrome. 6 Thus, there is need to find novel or repositioned antimycobacterial agents with unique modes of action and improved safety profiles. 7
Natural products have been a source of antimicrobial agents, with several frontline drugs originating or possessing templates from microbial and plant sources. 8 Kigelia africana (Lam.) Benth. (Synonym: Kigelia pinnata, the so called ‘‘sausage tree’’), is a member of Bignoniaceae family and the sole species in its genus. Its roots, bark, fruits, flowers, leaves and seeds are utilized in African and Asian traditional formularies to manage skin infections, infertility, gynecological complaints, venereal diseases, hypertension, rheumatism, toothache, malaria, cough, asthma and TB.9–11 Bioactivity studies have revealed that extracts from this species has antimicrobial, antioxidant, anti-inflammatory, analgesic, antimalarial, hepatoprotective, antiulcerogenic, antidiabetic and anticancer activities.12,13 These bioactivities are known to be due to the presence of phytochemicals such as phenolics, iridoids and limonoids, 13 naphthoquinones, phenyl ethanoglycosides, terpenes, terpenoids, lignans and steroids.14,15
The stem bark of K. africana is utilized for TB treatment,9,10,16 but studies targeting the isolation and evaluation of antitubercular compounds from this species are limited. Only one study by Wadhwani et al 17 isolated nine phytochemicals (cycloolivil, cluytyl ferulate, dehydro-α-lapachone, D-sesamin, kigelinone, lapachol, paulownin, tecomaquinone-I, and wodeshiol) from the heartwood of K. africana followed by in silico analyses. The authors reported that tecomaquinone-I was the only compound that exhibited potent antitubercular activity.
In the present study, in silico analyses were performed to investigate the potential antimycobacterial activity of known compounds isolated from K. africana stem bark against key M. tuberculosis, Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus) targets. Through prediction of the binding affinities and interaction profiles, the in silico approach provides results that support the development of K. africana-derived scaffolds as promising candidates for novel and repurposed antitubercular drugs.
Methods
Plant Material
Stem bark of K. africana was collected from Bupeni village, Kaliro District, Eastern Uganda in March 2019. The plant was identified by Mr. Protase Rwaburindore, a botanist at Makerere University Herbarium where a voucher sample of the stem bark (no. IG2019/002) was deposited. The bark was shade-dried for two weeks, and ground using a blender, yielding 1 kg of a brownish-red powder.
Extraction and Isolation of Pure Compounds
The dichloromethane/methanol (1:1) crude extract was obtained by maceration for 24 h. The extraction was repeated twice, and the pooled extract concentrated by rotary evaporation. The crude extract (150 g) was divided into two portions: one half was stored in a refrigerator and the other was subjected to chromatographic fractionation.
Briefly, 200 g of silica gel (70-230 mesh size) was used for column chromatography, using hexane as the mobile phase, and increasing polarity using ethyl acetate. Different fractions collected were spotted on TLC plates, and those with similar TLC profiles were combined. Fraction I eluted with 2% ethyl acetate in hexane yielded an amorphous solid (compound
Spectroscopic Analysis of Compounds
The isolated compounds were subjected to NMR spectroscopy on a Bruker AV-500 spectrometer, that is, 1D (1H and 13C) and 2D (1H-1H COSY, HSQC and HMBC) analysis of the samples dissolved in deuterated chloroform. The spectra in FID format were processed using MestReNova (version 8.1.1). The residual chloroform peaks were used as references. 18
Protein Preparation
The Autodock 4 (1.5.7) was selected and utilized to prepare the Protein Data Bank (PDB) file for the proteins of M. tuberculosis (PDB ID: 4QIJ and 5HKF), 19 E. coli (PDB ID: 5L3J and 8GZY) and S. aureus (PDB ID: 2QIL).20,21 The structural integrity was modified by removing the water molecules, ions, the Kollman charges were added and the other cofactors, and then followed by addition of the polar hydrogen atoms to the system. The protein structures were subsequently saved in PDBQT format. To realign all the structural loops and determine the binding active sites, BIOVIA Discovery Studio v.2025 Visualizer software was used. 22
Ligand Preparation
Information on the antibacterial potential of K. africana constituents against E. coli and S. aureus was obtained from previous studies,23,24 and the phytochemicals selected for this study were those characterized (
Drug Likeness Properties and ADME Analysis of the Phytochemicals
The SMILES format of the compounds was pasted onto ADMETLAB3.0 (https://admetlab3.scbdd.com/documentation) and SWISSADME (https://www.swissadme.ch/index.php) to generate their pharmacokinetic properties/ADMET and drug-likeness parameters.26,27 The Bioavailability Radar tool was used to determine the oral bioavailability of the compounds.28,29
Molecular Docking
Molecular docking was done using AutoDock Vina (1.1.2) with automation using Shell script, a configuration file containing the grid parameters and center values obtained from AutoDock 4 were passed as parameters.30,31 The binding energies (kcal/mol) were obtained together with the output pdbqt file. 32 Vina_Split software was used to separate the output file into individual poses for the ligands. BIOVIA Discovery Studio was used to analyse the interactions and generate the 2D and 3D structures of the best poses with minimal energies determined by AutoDock.
Toxicity Prediction
Toxicity of the compounds was predicted using the ProTox-II web server (https://tox.charite.de/protox3/), an in silico platform for computational toxicology. The chemical structures, in SMILES format, were submitted to the server to predict a wide range of toxicological endpoints. Predictions were based on an integrated methodology that includes molecular similarity, pharmacophore-based analyses, and machine learning models trained on publicly available toxicological data. The endpoints evaluated included hepatotoxicity, respiratory toxicity, cardiotoxicity (hERG inhibition), mutagenicity (Ames test), and organ-specific toxicities, each reported with an associated probability score. The acute oral toxicity (LD50) of each compound was predicted. 33
Statistical Analysis
Molecular docking results were analyzed descriptively based on the binding affinities (kcal/mol) and the visualized interactions of ligands with target proteins. Binding affinities of the test ligands were compared to those of reference antitubercular drugs (isoniazid and ethionamide). Drug-likeness and ADMET properties were interpreted according to standard rules (Lipinski, Ghose, Veber, Egan, Muegge). The predicted LD50 values of the isolated compounds were classified according to the Globally Harmonized System (GHS). 33 Due to the computational nature of the study, no further inferential statistical analyses were performed.
Results
Isolated Compounds from K. africana Stem Bark
Four compounds were isolated from the stem bark of K. africana (Figure 1). They were conclusively identified as demethylkigelin (

Structure of Compounds Isolated from Dichloromethane/Methanol Extract of K. africana Stem Bark: Demethylkigelin (
NMR Spectral Data (1H NMR, CDCl3 500 MHz and 13C NMR, 125 MHz) of Compound
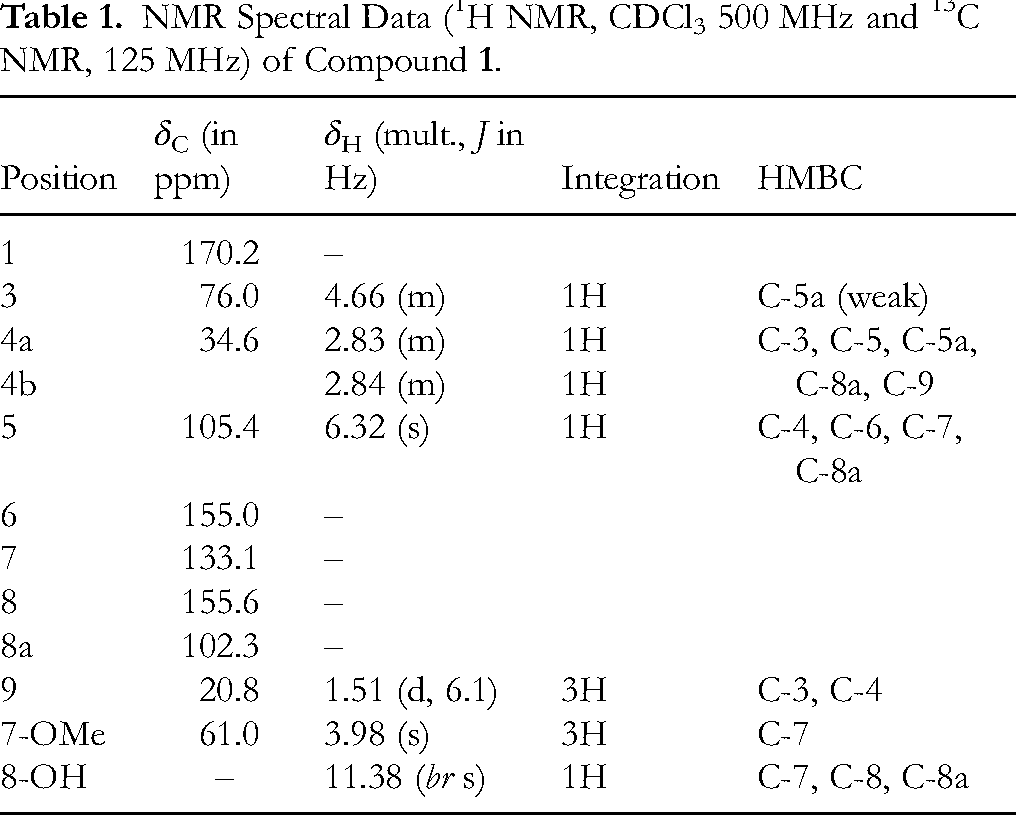
The presence of a signal at δC 170.2 in the 13C NMR spectrum, was suggestive of a lactone moiety. The 1H NMR spectrum (Figure S3) exhibited broad singlet at δH 11.38, attributed to the 8-OH group involved in an intermolecular hydrogen bond with the lactonyl carbon oxygen (C-1). Both the 1H NMR and the 1H-1H COSY spectra (Figure S4) revealed the presence of 2-hydroxy-propyl moiety of a typical cyclic ester [δH 1.51, d (J = 6.1 Hz, 3H, H-9), 2.83-2.84, m (2H, H-4a & H-4b), 4.66, m (1H, H-3)] whose attachment at C-5a of the aromatic ring was based on the HMBC correlation (Figure S5) between H-4a, 4b with carbons resonating at δC 102.3 (C-8a), 105.6 (C-5) and 135.6 (C-5a). These partial structures were joined to give the final structure of compound
Compound
NMR Spectral Data (1H NMR, CDCl3 500 MHz and 13C NMR, 125 MHz) of Compound

Compound
NMR Spectral Data (1H NMR, CDCl3 500 MHz and 13C NMR

Compound
NMR Spectral Data (1H NMR, CDCl3 500 MHz and 13C NMR, 125 MHz) of Compound

The compound also showed protons at δH 3.53, 5.15, and 5.35, suggesting the presence of three protons corresponding to that of a tri-substituted and a di-substituted olefinic bond. The proton corresponding to the H-3 of a sterol moiety appeared as a triplet of doublet of doublets at δH 3.52. The above spectral data supported the presence of a sterol skeleton having a hydroxyl group at C-3 position with two double bonds at C-5/C-6 and C-22/C-23 with six methyl groups which was supported by the key COSY (Figure S18) and HMBC (Figure S19) correlations. The 1H and 13C NMR values for all the protons and carbons (Table 4) were assigned on the basis of COSY, HSQC (Figure S20) and HMBC correlations.
Molecular Docking Results
The interactions between ligands of the compounds and specific target protein residues by molecular docking simulation were investigated to validate the antitubercular and antibacterial efficacy of the isolated compounds
Docking Score Results of M. tuberculosis, E. coli and S. aureus Receptors with Potential Inhibitors.
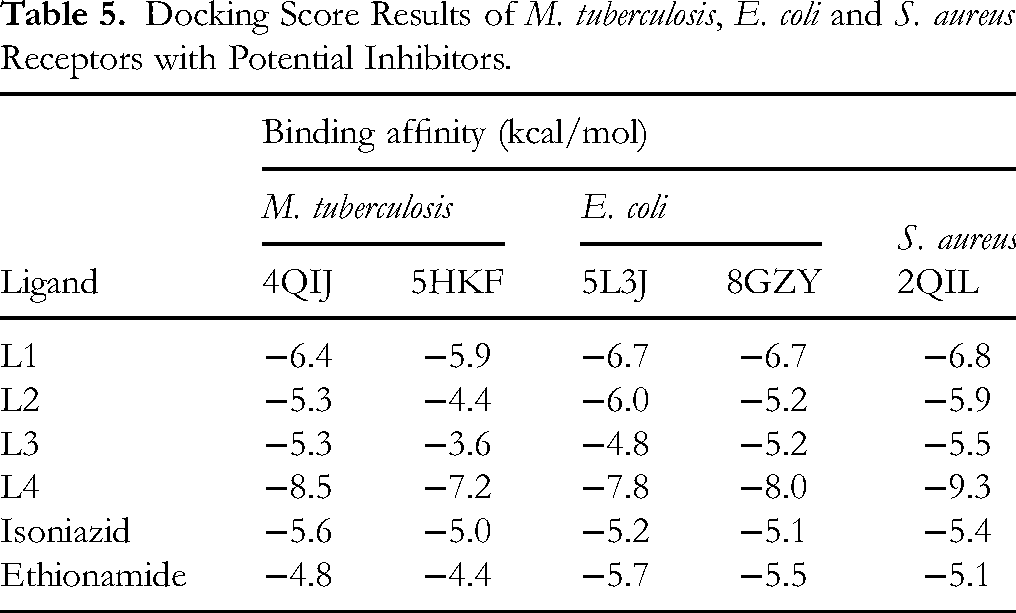
Ligand 4 (L4) demonstrated a strong binding affinity (binding score of −8.5 kcal/mol) in comparison to −5.9, −4.4 and −3.6 kcal/mol of L1 to L3 against 4QIJ, a protein target for M. tuberculosis. Similarly, it had the strongest binding affinity (binding score of −7.2 kcal/mol) against 5HKF, another protein target for M. tuberculosis. The same ligand (L4) demonstrated strong binding affinities (binding scores of −7.8 and −8.0 kcal/mol, respectively) against 5L3J and 8GZY, which are protein targets for E. coli. Interestingly, the same ligand had the best inhibitory activity with the highest binding affinity (binding score of −9.3 kcal/mol) against 2QIL, the protein target for S. aureus.
Pharmacokinetic Studies Results
The pharmacokinetic and physicochemical characteristics of the compounds (lipophilicity, blood-brain barrier (BBB) penetration potential, and interactions with various cytochrome enzymes) were evaluated (Table S1). These characteristics are essential for determining the compounds’ overall drug-likeness, potential adverse effects, and bioavailability, all of which are important factors in drug discovery and development. The bioavailability scores of the four compounds ranged from 0 to 0.56. All the compounds stand out for having zero violations of the Lipinski, Ghose, Veber, Egan and Muegge rules.
Toxicity Prediction Results
In order to predict the potential toxicity of the compounds, a computational toxicology analysis was performed using the ProTox-II web server. This in silico platform integrates various computational methods (such as molecular similarity searches, fragment-based analysis, and machine learning models) to predict a wide range of toxicological endpoints. Each compound was evaluated for acute oral toxicity, hepatotoxicity, respiratory toxicity and cardiotoxicity, with corresponding probabilities provided to indicate the reliability of each prediction. The results showed that compounds 1 and 4 could cause respiratory toxicity, with the former possibly causing cardiotoxicity (Table 6).
In silico Predicted Toxicities of the Compounds Isolated from K. africana.

Discussion
Kigelia africana, a member of the trumpet creeper or catalpa family is a heavily exploited medicinal plant for its medicinal values. In the present study, chromatographic fractionation of its dichloromethane/methanol stem bark extract followed by extensive structural elucidation yielded four compounds (
Extensive comparison of spectral data of compound
The 13C NMR signal at δC 174.0 ppm indicated the presence of an ester carbonyl, which was further substantiated by the HMBC correlations between the methylene protons of the butyrate chain and the carbonyl carbon. Together, these spectral data with the terminal methyl group signal at δH 0.90 (t, J = 6.9 Hz) confirmed the attachment of a butyrate group, completing the structure assignment. Compound
Compound
Compound
In molecular docking studies, the binding interaction achieved for ligand-protein targets provide good insights into their corresponding shapes and electrostatic relationships.
47
In other words, docking score indicates the total energy realized during the interaction, with lower negative values signaling stronger interactions (see Table S1). The binding results of compounds
In the pharmacokinetic studies, all the compounds stood out for having zero violations of the Lipinski, Ghose, Veber, Egan and Muegge rules. This indicates that the compounds are compliant with established drug-likeness criteria.
51
Furthermore, the drug's permeability and oral bioavailability depend on the values of topological polar surface area (TPSA) and molecular lipophilicity potential (log P). In this context, log P indicates the compound's distribution coefficient between n-octanol and water, and it plays a vital role in the cellular membrane's interactions with other proteins.52,53 All the compounds exhibited log P values above five, except compound
During drug metabolism, there are essential enzymes under Cytochrome P450 (CYP) family which are involved in the phase 1 metabolism of xenobiotics. It is known that some phytochemicals isolated from different sources have the ability to inhibit the CYP isoforms, leading to drug-drug interactions.
54
In the present study, it was predicted that compounds
Computational toxicity predictions showed that L1 (demethylkigelin) exhibited a moderate acute oral toxicity with a predicted LD50 of 500 mg/kg (toxicity class 4 according to GHS criteria). This compound was also predicted to be active for both respiratory and cardiotoxicity, although with lower probabilities for the latter, indicating potential adverse effects on these systems. In contrast, L2 (tyrosyl butyrate) was the safest compound, with a high predicted LD50 of 5000 mg/kg, classifying it as non-toxic (class 5). The high probability scores for its predicted inactivity across all the tested endpoints further support its low toxicity profile. On the other hand, L3 (stearic acid) had a relatively low acute oral toxicity (LD50 of 900 mg/kg) and was predicted to be inactive for all the other tested endpoints, with a very high confidence score for its inactivity in cardiotoxicity. Stigmasterol (L4) had a similar LD50 to L3 (stearic acid), but was predicted to be active for respiratory toxicity with a high probability. Overall, the results suggested that while L2 and L3 showed promising low toxicity profiles, L1 and L4 warrant further investigation, especially for their potential respiratory and cardiotoxic effects.
Despite these interesting results, the present study has some limitations. The study was largely in silico, relying on molecular docking and ADMET predictions. These methods provide useful preliminary insights into drug-target interactions, pharmacokinetic and toxicity properties but they do not substitute for experimental validation. Further, we did not extend the analysis to KEGG pathway or STRING network enrichment, as these require a broad and validated target set to generate meaningful mechanistic insights. Future studies integrating experimental activity data with target prediction will allow more reliable pathway and network analyses. The predicted binding affinities, ADMET and toxicity properties may differ under physiological conditions, and therefore, in vitro and in vivo studies are required to confirm the antimycobacterial activity, safety, and pharmacological potential of the compounds identified from K. africana stem bark.
Conclusion
This study found that K. africana stem bark possesses bioactive compounds that are potential inhibitors of M. tuberculosis with good to better binding affinities and stable interactions. We recommend that future studies should perform in vitro and in vivo validation of the bioactivity and toxicity of the characterized compounds.
Supplemental Material
sj-docx-1-npx-10.1177_1934578X251388844 - Supplemental material for In silico Antimycobacterial Evaluation of Compounds Isolated from Kigelia africana Stem Bark
Supplemental material, sj-docx-1-npx-10.1177_1934578X251388844 for In silico Antimycobacterial Evaluation of Compounds Isolated from Kigelia africana Stem Bark by Ivan Gumula, Mary Achiro, Sarah Kiwanuka Nanyonga, Denis Akampurira, Patrick Onen, Ronnie Tumwesigye and Timothy Omara in Natural Product Communications
Footnotes
Acknowledgments
The authors are grateful to the Institute of Chemistry, University of Potsdam, Germany for NMR analysis of the isolated compounds.
Ethical Approval
Ethical approval is not applicable for this article.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Statement of Human and Animal Rights
Not applicable for this article.
Author Contributions
Conceptualization: Ivan Gumula, Mary Achiro, Patrick Onen; Methodology: Ivan Gumula, Sarah Kiwanuka Nanyonga, Patrick Onen, Ronnie Tumwesigye, Timothy Omara; Investigation: Mary Achiro, Patrick Onen, Ronnie Tumwesigye; Formal analysis: Mary Achiro, Patrick Onen, Ronnie Tumwesigye; Writing—original draft: Ivan Gumula, Timothy Omara; Writing—review and editing: Ivan Gumula, Timothy Omara. All authors read and approved the final manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Kyambogo University through the Competitive Research Grant awarded to the first author.
Declaration of Conflicting Interest
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors declare no financial or personal conflicts of interest. Timothy Omara discloses his role as a member of the Editorial Review Board of Natural Product Communications. He had no involvement in the editorial decision making for this article.
Data Availability
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.