
Abstract
Objective
Birth prevalence of Cobalamin (Cbl) C or D defects in Portugal is an estimated 1:85,000, one of the highest worldwide. We compared the genotype/phenotype of patients identified with CblC or CblD before and after the implementation of expanded newborn screening.
Methods
Twenty-five Portuguese CblC/D patients, 14 symptomatic and 11 identified through screening, were diagnosed using gas chromatography or tandem mass spectrometry. Molecular characterization was performed through the study of MMACHC and MMADHC genes.
Results
The most common MMACHC mutation, c.271dupA, was present in 100% of MMACHC alleles of all CblC screened patients, in contrast with the 61% identified before expanded newborn screening. All studied cases (except one, who presented a CblD deficiency) presented a CblC defect. More CblC late-onset patients were diagnosed before the introduction of newborn screening than in the post newborn screening era, probably because some early onset patients died without a definitive diagnosis.
Conclusion
The molecular data found in this cohort contribute to the improvement of screening and diagnosis of Cbl defects and would enable a confirmatory diagnosis of these patients, reducing the need for complex, costly, laborious, and time-consuming biochemical/enzymatic tests.
Introduction
Acquired from the diet, cobalamin (Cbl) or vitamin B12 is metabolized within the cell into methylcobalamin and adenosylcobalamin, 1 essential coenzymes to methionine synthase and methylmalonyl-CoA mutase. Inborn errors of intracellular cobalamin metabolism, resulting from impaired conversion of dietary vitamin B12 to its two metabolically active forms, cause methylmalonic aciduria (MMA) and homocystinuria (HC), and a decrease of methionine.2,3 In Portugal, only CblC and CblD deficiencies have been detected.
CblC (MIM# 277400), the most common inherited disorder of vitamin B12 metabolism, has two distinct phenotypes: (i) early onset (EO), a multisystemic disease that presents with MMA and HC, and clinical symptoms such as feeding difficulties, hypotonia, hydrocephalus, progressive developmental delay, seizures, mild dysmorphic features, anaemia, and haemolitic uremic syndrome appearing within the first year of life, and (ii) late onset (LO), which presents later in life, with a relatively milder clinical phenotype, progressive neurological symptoms, and behavioural disturbances. 4 The treatment of both forms with hydroxycobalamin, betaine, and folic acid usually decreases metabolite levels. 2 Early treatment is clearly advantageous for patients with LO CblC defect, and for those with EO CblC treatment improves both survival and non-neurological symptoms, but its effect on neurocognitive development remains controversial. 5
CblC defects are caused by mutations in the MMACHC gene (ID: 25974; MIM# 609831), located in the 1p34.1 locus. 6 Three common mutations have been identified: c.271dupA, c.394C > T, and c.331C > T.6–8 The c.271dupA and c.331C > T mutations have been associated with EO disease, and c.394C > T primarily with LO disease. Some MMACHC mutations have been observed to correlate with age of onset and others cluster according to ethnicity, probably as a result of different founder effects.6,7,9 MMACHC is responsible for early processing of both cyanocobalamin (decyanation) and alkylcobalamins (dealkylation) in mammalian cells. 10
Patients with the CblD genetic defect (MIM# 277410) can have three distinct biochemical phenotypes: (i) isolated homocystinuria (CblD-HC), (ii) isolated methylmalonic aciduria (CblD-MMA), and (iii) combined homocystinuria and methylmalonic aciduria (CblD-MMA/HC). 11 CblD-HC patients present with developmental delay, ataxia, and megaloblastic anaemia; CblD-MMA patients show respiratory distress, cranial haemorrhage, seizures, and an abnormal electroencephalogram; CblD-MMA/HC patients present with developmental delay, seizures, hypotonia, lethargy, and megaloblastic anaemia.11,12 Regardless of their clinical phenotype, patients with CblD defects usually respond well to betaine, folic acid, and OH-cobalamin treatment, even though they remain neurologically impaired. MMADHC (MIM 611935) is the gene (chromosome region 2q23.2) 12 responsible for the CblD defect of vitamin B12 metabolism. Usually, individuals with CblD-HC carry missense mutations causing amino acid substitutions towards the C-terminal part of the protein, and patients with CblD-MMA carry mutations causing a premature stop of translation towards the N-terminal part of the protein, on at least one allele. The combined phenotype (CblD-MMA/HC) is associated with deleterious nonsense and missense mutations in the middle of the protein. 12 Recent studies demonstrate that MMADHC interacts directly with MMACHC, and the MMACHC–MMADHC complex is involved in Vitamin B12 trafficking. 13 Genotype/phenotype correlation analysis of the three CblD variants suggests that the N- and C-termini of MMADHC have specific functions in the mitochondrion and cytoplasm, respectively.14,15
Our molecular findings on CblC/D Portuguese patients indicate the type of patients who may be detected in the neonatal period. While there is no good published evidence of the benefits of screening for CblC/D defects, there is a great deal of anecdotal evidence suggesting that some EO patients do much better after early diagnosis. 16 Improving the newborn screening diagnosis therefore seems worthwhile.
Methods
Of the 25 Portuguese CblC/D patients in our study (12 males, 13 females), 14 symptomatic patients were identified up to 2004 by urinary organic acids and plasmatic amino acid analysis (group 1). The remaining 11 patients were identified after 2004, when expanded newborn screening was established (group 2), and were detected through acylcarnitines and amino acids analysis in dried blood spots. The age at diagnosis ranged between 25 days and 16 years for group 1, and 3–7 days, for group 2. Informed consent was obtained from all patients included in this study.
Two API2000 triple quadrupole tandem mass spectrometers (ABSciex) were used to perform routine MS/MS neonatal screening, including the analysis of amino acids and acylcarnitines as butyl esters. 17 Screening for CblC/D was included in the Portuguese Newborn Screening Program in 2004 and has been performed at national level since 2006. It is based on the detection of a high propionyl/acetylcarnitine ratio (C3/C2; normal <0.25), alongside elevated levels of propionylcarnitine (C3; normal <6.3 µM) and an increased propionylcarnitine/methionine ratio (C3/Met; normal <0.3), as well as normal to low levels of methionine (Met; normal >7.0 µM). More recently, the cut-off value of C3 metabolite was modified to 5.3 µM and new markers were introduced: propionylcarnitine/free carnitine (C3/C0; normal <0.4) and methionine/phenylalanine (Met/Phe; normal >0.3).
Urinary organic acids were extracted by a standard ethylacetate procedure, derivatized with Bis-trimethylsilyltrifluoroacetamide and pyridine, and analysed on a gas chromatography–mass spectrometry system (GC/MS Shimadzu QP 2010 Plus). Plasma amino acid analysis was performed by ion-exchange chromatography (Biochrom 30 Gomensoro Instrumentation Cientifica).
Genomic DNA was extracted from peripheral blood; where patients were already deceased, DNA was extracted from Guthrie cards, using standard procedures. The coding exons and exon–intron boundaries of MMACHC and MMADHC genes were amplified by polymerase chain reaction as previously described,6,12 and the products were sequenced using the BigDye Terminator Cycle Sequencing Version 3.1 (Applied Biosystems, Foster City, CA), and analysed on an ABI 3130XL DNA Analyser.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975 (revised 2000).
Results
Over the past 25 years, our centre (National Institute of Health, Porto, Portugal) has been involved in CblC/D defect diagnosis and newborn screening. Diagnosis in the pre-newborn screening era was based on the identification of urinary and circulating metabolites and, whenever possible, confirmed by either fibroblast complementation assays in the early stages of the programme 18 or, more recently, by molecular studies.6,11 Based on the results of 10 years of expanded newborn screening, the birth prevalence of CblC/D defects in Portugal is 1:85,000, one of the highest known prevalence in the world. 19
Molecular and biochemical data of CblC patients, diagnosed in pre newborn screening era.
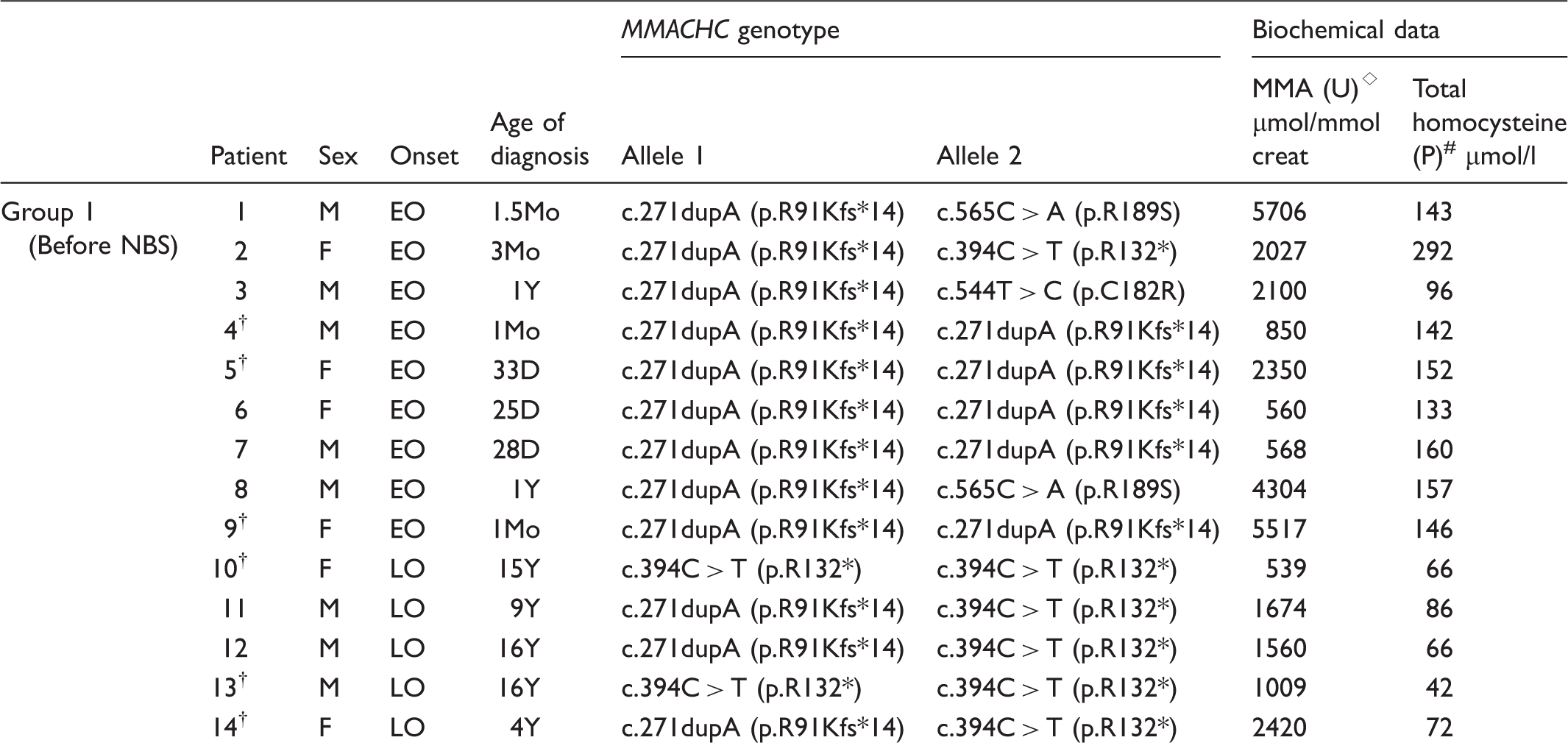
creat: creatinine; D: days; EO: early onset; F: female; LO: late onset; M: male; †: died (patients 4, 5, 9, and 14 died at diagnosis time; patients 10 and 13 died at 21 and 26 years old, respectively); MMA: methylmalonic acid; Mo: months; P: plasma; U: urine; Y: years; ⋄: MMA reference values: 0–1 Mo <15.6 µmol/mmol creat, 1Mo–5Y <6.2 µmol/mmol creat, 5Y–18Y <3.3 µmol/mmol creat; #: total homocysteine reference values: 4.0–14.0 µmol/l; *: stop codon.
Molecular and biochemical data of CblC/D patients, diagnosed in newborn screening era.
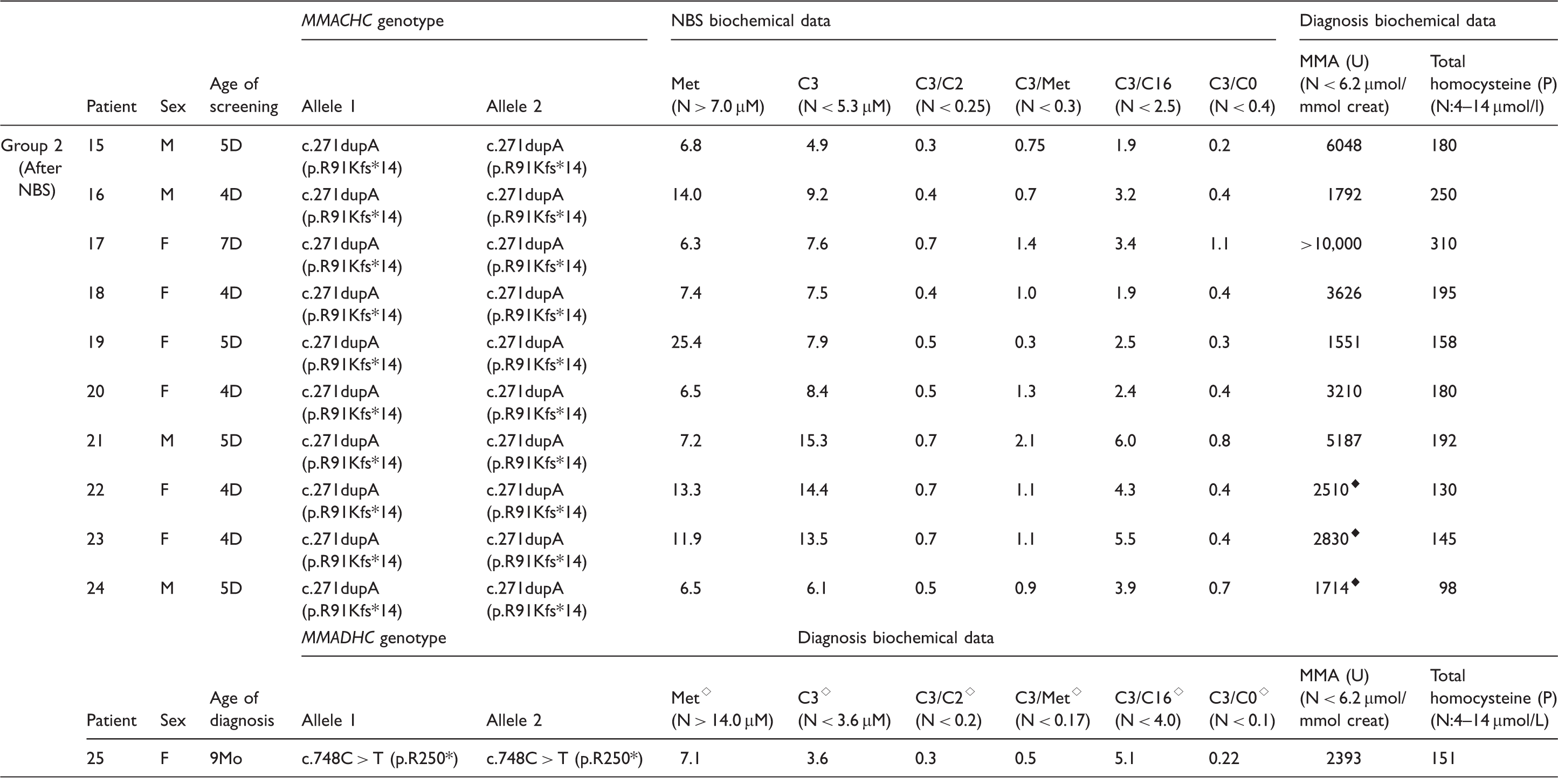
C0: free carnitine; C2: acetylcarnitine; C3: propionylcarnitine; C16: palmitoylcarnitine; D: days; F: female; M: male; Met: methionine; Mo: months; ⋄: reference values: 1Mo–1Y.
The c.271dupA mutation was the most common MMACHC disease allele (77%) in our study. Mutations c.394C > T (p.R132X), c.544T > C (p.C182R), and c.565C > A (p.R189S) were found in 16, 2, and 4% of the total MMACHC alleles, respectively. As previously noted, the c.271dupA, c.544T > C, and c.565C > A mutations were associated with EO disease, while the c.394C > T mutation underlies LO forms of the disease.
Discussion
Defects in the MMACHC gene are the major cause of combined MMA and HC among Southern European (Portugal, Spain, and Italy) 7 and Chinese patients. 20 The 14 diagnosed symptomatic patients in our study demonstrated significant molecular and clinical heterogeneity, with different genotypes and both EO and LO clinical forms. However, since the implementation of expanded newborn screening in 2004, all CblC defect patients we have identified share the same genotype: the c.271dupA mutation in a homozygous state. These data confirm that newborn screening allows the detection of the most severe EO cases. No LO patients were identified by newborn screening during this period. This may be related with the low birth prevalence of LO forms, but these patients are more difficult to identify by newborn screening. To ensure that all cases are identified we could decrease the cut-off markers, but this would substantially increase the number of false positives, the cost of the newborn screening for these diseases, and also parental anxiety. The implementation of second-tier tests for total homocysteine and methylmalonic acid determination may help to overcome this problem and allow the diagnosis of LO cases in our population. Another study has suggested that the C3/C2 ratio (and perhaps other ratios such as C3/C0) should be used as markers for disorders of propionate metabolism when the total carnitine level is low, because it may lead to a lower C3 acylcarnitine level. Therefore, depending on screening cut-offs, CblC patients with LO forms, mild mutations, and low total carnitine levels could be missed in newborn screening. 21
Our study allowed for the recognition of strong genotype/phenotype correlations, mainly in CblC defects, as already outlined in a large French-Canadian study. 6 When a correlation to the clinical phenotype was attempted, homozygosity for c.271dupA appears to be uniformly associated with an EO phenotype. In particular, we found that in the pre-neonatal screening era, 61% of EO alleles harboured the c.271dupA (five patients of this group homozygous for this mutation), whereas the c.394C > T mutation accounted only for 4% of EO alleles. In the LO cases diagnosed in the pre-neonatal screening era, these frequencies were quite different: the c.271dupA and the c.394C > T mutations were found in 30 and 70% of the alleles, respectively. The latter mutation was never detected in homozygosity in EO patients, whereas it was found in compound heterozygosity with c.271dupA in one case with early disease, and in three LO cases. In the post-newborn screening era, all CblC defects presented the c.271dupA mutation in a homozygous state. The reduced number of EO patients identified in the pre-newborn screening era was probably due to the patients’ death without a definitive diagnosis.
These genotype/phenotype observations are consistent with the clinical heterogeneity observed in CblC disease. 22 While age at onset has been used to subcategorize CblC individuals into EO and LO groups, individuals with LO CblC disease may have had disease-related symptoms, which went unrecognized, before they were diagnosed. 21
Molecular strategies based on the simultaneous detection of the two most common mutations (i.e. c.271dupA (EO) and c.394C > T (LO)) would be advantageous as they might characterize almost the whole set of Portuguese patients, enabling a cost- and time-effective confirmatory diagnosis, and reducing the need for more laborious and time-consuming biochemical testing in cultured cells. The two other mutations found in our study suggest that the likelihood of detecting new Southern European variants in MMACHC gene is low. A quick testing for the most common c.271dupA mutation allowed the rapid confirmation of the MMACHC infants detected by expanded newborn screening, as all were homozygous for this mutation. This genotype is associated with the most severe phenotype of EO presentation and contributed to the high prevalence of EO patients detected by newborn screening compared with that among patients identified in the pre-newborn screening era, already in a symptomatic stage of the disease. This may be explained by the death, without a definitive diagnosis, of patients with severe EO forms in early infancy.
We detected one patient, in the post-newborn screening era, who presented with a CblD defect. This case, diagnosed at age nine months, was a neonatal screening false negative, because the C3 cut-off at that time was higher than that currently in use (6.3 µM instead of 5.3 µM). This patient presented with EO, but not a severe form of the disease, thus corroborating our hypothesis that milder EO and LO forms of CblC/D disease may not be identified at newborn screening. Additionally, during the newborn screening era, we have identified secondary cobalamin defects in two neonates of vegetarian mothers, and two other children whose mothers had pernicious anaemia.
Conclusion
Comparison of the two groups in our cohort of 25 ClbC/D cases demonstrates that LO forms were more frequent in the pre-newborn screening era, probably because many EO patients died without a definitive diagnosis. Clinicians should be aware that LO patients may not be detected by newborn screening. To avoid newborn screening false positives and false negatives for ClbC/D defects, we are now implementing second tier tests that will allow the detection of the increased levels of total homocysteine and methylmalonic acid, and lower C3/C2 and C3 cut-off levels, respectively. Our data corroborate the importance of molecular genetic testing for an accurate genetic counselling and for a confirmatory diagnosis of CblC/D, thus reducing the need for complex, costly, laborious, and time-consuming biochemical/enzymatic tests.
Footnotes
Acknowledgements
We are grateful to the European Network and Registry for Homocystinurias and Methylation Defects – EHOD project (N°2012_12_02) for the partial support of this work. We are also grateful to Dr Francisca Coutinho for editorial assistance and to clinicians and patients.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.