
Abstract
Posttransplant lymphoproliferative disorder (PTLD) is a rare lymphoid and/or plasmocytic proliferation that occurs after allogeneic hematopoietic stem cell transplantation (allo-HSCT). We aimed to identify the pathologic features and clinical outcomes of T-cell PTLD, an extremely rare subtype of PTLD, after allo-HSCT. In this study, six allo-HSCT recipients with T-cell PTLD from five transplant centers in China were enrolled. All the T-cell PTLD were donor-derived, and three patients were with monomorphic and three with polymorphic types, respectively. All patients received cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP)-based chemotherapy. Five patients achieved complete response (CR), and one experienced progressive disease (PD). The median time from HSCT to onset was 4 (range: 0.6-72) months, analyzed in combination with the other 16 patients with T-cell PTLD identified from previous reports. About 56.3% of the T-cell samples (9/16) were positive for in situ hybridization with an Epstein–Barr virus (EBV)-encoded small nuclear early region (EBER ISH). CHOP-based chemotherapy might be the optimal strategy for patients who showed no response to empiric therapy with a CR rate of 87.5%. In conclusion, our study observed that T-cell PTLD has distinct clinical manifestations and morphological features, which characterized by less relation to EBV, later occurrence, and poorer prognosis when compared with B-cell PTLD.
Keywords
Introduction
Posttransplant lymphoproliferative disorder (PTLD) is a life-threatening complication of allogeneic hematopoietic stem cell transplantation (allo-HSCT), with an incidence ranging from 3% to 9%1,2. For the patients with inappropriate therapy, the mortality could be as high as 52.7% within 3 years 3 . Most PTLDs after allo-HSCT are originated from donor-derived B-cell, associating with Epstein–Barr virus (EBV) reactivation4,5. In the last two decades, risk-stratified therapeutic strategies 5 , particularly CD20 antibody, have greatly improved the survival of B-cell PTLD patients 3 . About 61.8% to 69.4% patients achieved complete remission (CR) after rituximab-based therapy3,6.
T-cell PTLD is extremely rare after allo-HSCT. Kuno et al. 4 reported that T-cell PTLD occurred in 0.39% of allo-HSCT recipients and was more likely to be EBV-negative and recipient-origin. The treatment of T-cell PTLD was different from B-cell PTLD. Because of its scarcity, knowledge about pathogenesis, risk factors and prognosis rely predominantly on case reports and small series, and its characteristics and therapeutic protocols were still unclear. In this case series study, we enrolled six cases of T-cell PTLD and aimed to identify their clinical characteristics and outcomes. In addition, we systemic reviewed 16 cases reported in previous studies.
Materials and Methods
Patient Selection
This multicenter, retrospective study was designed by the First Affiliated Hospital of Soochow University, Wuhan Tongji Hospital, the First Affiliated Hospital, Zhejiang University School of Medicine, Shanghai General Hospital, and Shanghai Ruijin Hospital. Allo-HSCT recipients who transplanted between January 2017 and September 2023 were retrospectively screened, and the eligibility criteria were as followed: (1) aged ≥ 16 years; (2) diagnosed with T-cell PTLD. The patients without complete medical information were excluded. The last follow-up was December 31, 2023. The study was approved by the Institutional Review Board of each participated hospital and was conducted in accordance with the Declaration of Helsinki.
Morphological and Molecular Analysis
All biopsy materials were diagnosed by hematopathologists according to the World Health Organization Classification of Tumors of Hematopoietic and Lymphoid Tissue published in 2017 7 . Immunohistochemical (IHC) analysis was performed with monoclonal antibodies on formalin-fixed, paraffin-embedded (FFPE) tissues. In situ hybridization (ISH) with an EBV-encoded small nuclear early region (EBER)-1 probe was performed to detect possible EBV infection. EBV-DNA loads in peripheral blood were measured by quantitative polymerase chain reaction (qPCR).
A donor-recipient chimerism study of tumor tissue was performed by quantitative PCR with fluorochrome-labeled primers for polymorphic microsatellite markers containing short tandem repeat (STR) sequences or Y chromosome-based fluorescence in situ hybridization (FISH) analysis. FISH analysis of PTLD tissues was performed in cases after sex-mismatched allo-HSCT. To exclude the contamination of residual recipient-derived non-hematolymphoid cells, the FFPE tissues for the chimerism analyses were prepared after confirming that the tumor cells were numerically dominated by hematoxylin and eosin or IHC staining. For polymorphic PTLD, T-cell PTLD was diagnosed when T-cells account for the dominant clone of neoplastic cells.
Transplant Regimen
The preconditioning regimen included cytarabine, busulfan, cyclophosphamide, semustine, and anti-thymocyte globulin (ATG)8,9. The protocols for acute graft versus host disease (GvHD) and infection prophylaxis were reported previously 10 .
Definitions
Any regimens that include (1) ≥9.6 mg/kg intravenous busulfan, (2) ≥140 mg/m2 of melphalan were considered as myeloablative conditioning (MAC). EBV viremia was defined as any levels of EBV DNA detected in plasma by PCR, and the threshold for preemptive therapy was ≥1000 IU/mL.
Clinical staging of T-cell PTLD was evaluated according to the Lugano Modification of Ann Arbor Staging System, and the Lugano response criteria were introduced for response assessments 11 .
Data Collection
Investigators in each hospital utilized the institutional electronic medical records of clinical databases in each hospital. The collected data included information on patients’ demographics and diagnosis of the underlying disease, pathology, and immunohistochemistry of involved tissue. All the data were independently reviewed by two hematologists and one pathologist with rich experience.
Published Articles
We searched for all available articles in the PubMed database using the search terms “(T-cell) AND ([lymphoproliferative disorder] OR (PTLD)) AND (case report) AND (HSCT)” between 1990 to 2023, and reviewed case reports of T-cell PTLD after allogeneic HSCT.
Results
Clinical Features of Our Patients
Six patients with T-cell PTLD were identified, and their characteristics were shown in Table 1. The median age at allo-HSCT was 22 years old (range: 16–58). T-PTLD were diagnosed within 6 months post-HSCT, and the median interval from allo-HSCT to occurrence of symptoms was 53 (range: 20–114) days. All patients received MAC with ATG as aGvHD prophylaxis. Five patients received haplo-identical related donor (HID) HSCT, and experienced EBV viremia before PTLD with a median peak EBV-DNA load of 10.3 × 104 (range: 1.4–153 × 104) IU/ml. Case 4 was diagnosed with EBV-negative PTLD, as both the donor and recipient were negative for pre-transplant EBV serostatus 12 . Case 2 experienced EBV reactivation concurrent with aGvHD. All patients presented fever and lymphadenopathy when PTLD was diagnosed.
Patients’ Characteristics.

Pathological Findings of T-PTLD Patients
Three patients were diagnosed as monomorphic PTLD (PTCL-NOS, Peripheral T-Cell Lymphoma, not otherwise specified), and the other three were polymorphic diseases (Table 1). Donor-derived PTLD was verified in all six patients. IHC staining showed that four of the six patients had positive EBER ISH. The median Ki67 index was 60% (range: 30%–80%). T-cell PTLD neoplastic cells were positive for CD7 (3/6) and CD30 (3/6), and the majority tested negative for CD5 (4/6) and CD20 (4/6).
Representative IHC results of Case 1 (Polymorphic PTLD) are shown in Fig. 1. The neoplastic cells were positive for CD2, CD3, CD7, CD8, and CD43 (Fig. 1A–E), partially positive for CD79a, Granzyme B, and TIA-1 (Fig. 1F–H), and negative for CD5, CD20, and CD30. Scattered lymphocytes were positive for EREB (Fig. 1I). The Ki-67 of these cells was approximately 60% (Fig. 1J).
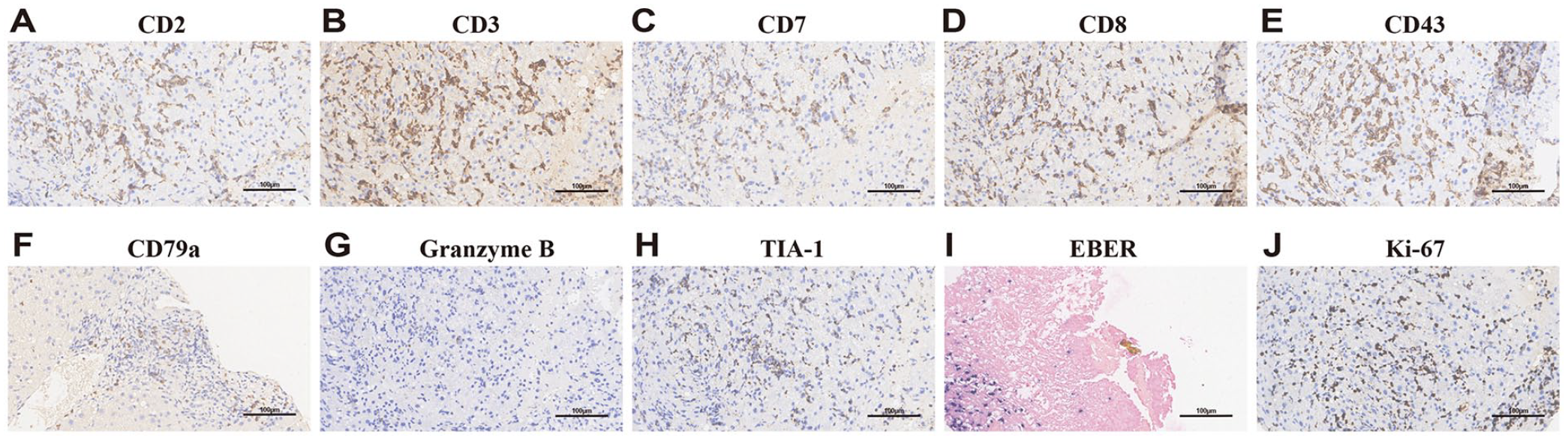
Representative histology and immunohistochemical analysis of T-cell PTLD (Case 1). Polymorphic PTLD, positive for CD2 (A), CD3 (B), CD7 (C), CD8 (D), and CD43 (E), partial positive for CD79a (F), Granzyme B (G), and TIA-1 (H) scattered positive for EBER ISH (I), Ki-67 (J) expression of neoplastic cells.
Treatment and Clinical Outcomes
Four patients received rituximab at 375 mg/m2 for pre-emptive therapy with a median of 2 (range: 1–4) doses for EBV viremia, and one patient turned negative for EBV DNA. It took a median of 20 (range: 4–36) days to diagnose T-cell PTLD from the detection of EBV viremia. Once T-cell PTLD was diagnosed with pathology, cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP)-based chemotherapy was initiated in all patients. Five patients achieved CR. In patients achieving CR, two died of infection and the other three were still in CR with follow-up. One patient experienced progressive disease (PD) and died of PTLD.
Clinicopathological Features of T-Cell PTLD After Allo-HSCT
We identified 16 patients with T-cell PTLD after allo-HSCT in literature review (Table 2)4,13–21. Among these 16 patients and 6 patients of our case series, the median age at transplantation was 32.5 (range: 2-67) years, and 36.4% patients (8/22) were 50 years or older at transplant. The primary diseases included myeloid malignancy (n = 8), lymphoid malignancy (n = 12), and aplastic anemia (n = 2). About 57.1% (n = 12) patients received HLA-matched allo-HSCT. Around 54.5% (n = 12) of patients received ATG as GvHD prophylaxis. About 68.2% (n = 15) of patients experienced aGvHD before PTLD. The median interval to PTLD was 4 (range: 0.6-72) months, and 72.7% (n = 16) occurred during the first year after allo-HSCT. About 50.0% of patients (n = 11) experienced EBV viremia before T-cell PTLD was diagnosed.
Literature Review of T-Cell PTLD Patients After Allo-HSCT.
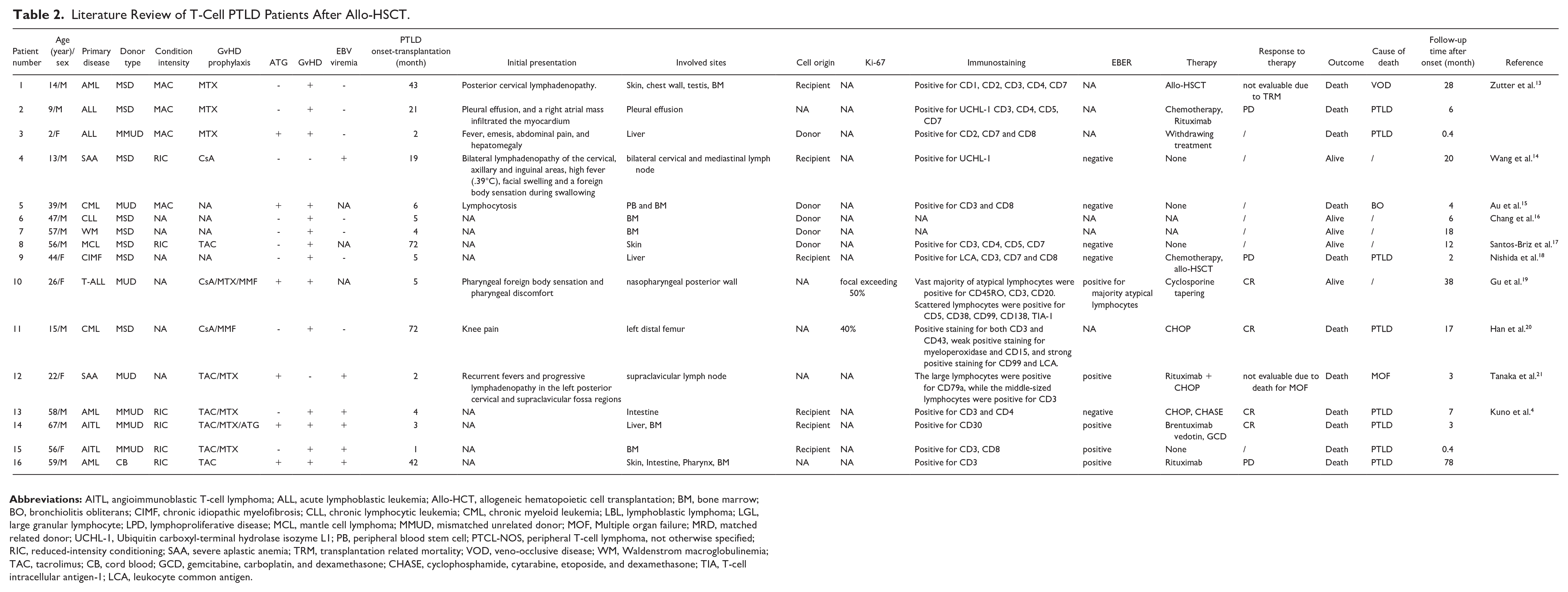
The most common symptom was fever, followed by pain or lump caused by lymphadenopathy. The lymph node was frequently involved, leading to the various clinical characteristics. The immunotypes of T-cell PTLD were heterogeneous. Of 16 patients with EBER ISH results, 56.3% of the tissues (9/16) were positive for EBER. Chimerism analysis showed that 64.7% of the cases (11/17) were donor-derived.
Sixteen of 22 T-cell PTLD patients received interventions, including chemotherapy (n = 13), allo-HSCT (n = 2) and one patient only with reduction of immunosuppressive agents. Of 15 patients in this study who had therapy information and response evaluation, 53.3% of the patients (8/15) received CHOP-based chemotherapy. 87.5% of these patients (7/8) achieved CR, and the CR rate of other therapies was 29% (2/7). About 40.1% of patients (9/22) died of PTLD. Kaplan–Meier analysis showed that the median OS of the 22 patients was 7 months.
Comparison of T-Cell PTLD and B-Cell PTLD
We further compared the features of T-cell and B-cell PTLD after allo-HSCT. The characteristics are summarized in Table 3. Few studies reported the incidence of T-cell PTLD after allo-HSCT, although T-cell PTLD accounted for approximately 5%–15% of all PTLD after solid organ transplantation (SOT)22,23. Several studies have provided dissociating patterns between EBV-positive and EBV-negative PTLD24–26. The relatively low positive rate of EBER ISH for T-cell PTLD compared with B-cell PTLD (53.3% vs. 60-80%4,5) is consistent with previous study 23 suggesting that the pathogenesis of T-cell PTLD may be different from B-cell PTLD. T-cell PTLD developed later than B-cell PTLD after allo-HSCT (median: 5 months vs. 2-3 months 27 ). Besides, the treatment of T-cell PTLD was more limited, and the OS of patients with T-cell PTLD was inferior to that of patients with B-cell PTLD (median OS: 6.5 months vs. 6.6 years) 28 , because B-cell PTLD would benefit from rituximab-based therapy 28 .
Comparison of T-Cell PTLD and B-Cell PTLD in Allo-HSCT Patient.

These data of T-cell PTLD were calculated from 22 patients in this study. Other data were summarized from systematic review of previous literatures.
Discussion
In this retrospective analysis, we presented the clinical features and outcomes of six patients with T-cell PTLD after allo-HSCT, and systemic reviewed 16 T-cell PTLD patients in the literatures. We found that T-cell PTLD had a later onset, weaker association with EBV, and a poorer response to chemotherapy compared with B-cell PTLD.
The impact of the intensity of conditioning regimen on the development of PTLD remains controversial. Previous studies revealed that reduced-intensity conditioning (RIC) with ATG as aGvHD prophylaxis increased the probability of EBV reactivation after allo-HSCT 29 , and RIC conditioning increased the risk of EBV-PTLD 30 . On the contrary, Xuan et al. 31 observed that intensified conditioning was one of the risk factors for EBV viremia and PTLD. However, T-cell PTLD is rarely reported. Tiede et al. 23 confirmed that allo-HSCT was associated with early-onset T-cell PTLD. The primary independent favorable prognostic factors included younger patients, T-cell PTLD of the large granular lymphocytic leukemia subtype, a combination of radiotherapy/radio chemotherapy and reduced immunosuppression 23 . In addition, patients receiving HID HSCT with ATG as GvHD prophylaxis were predisposing to higher incidence of EBV viremia (83.3% vs. 46%) and earlier onset of T-cell PTLD (median: 2 months vs. 5 months) in our series, compared with the other 16 T-cell PTLD patients.
EBV mainly infects B lymphocytes and has the capacity for transforming B-cells and driving the pathophysiology of B-cell PTLD32,33. As previously reported, less T-PTLD were EBV-related than that of B-cell PTLD 34 . The association between T-cell PTLD and EBV was weaker as a lower rate of EBER positivity in IHC was detected in this study. Nevertheless, Chuhjo et al. 35 found that EBV could infect T- and B-cells simultaneously, inducing clonal proliferation of both types of lymphocytes in profound immunocompromised patients. Some researchers believed that a cofounding, T-cell proliferation and transformation-promoting role of adjacent EBV-positive B-cells. In a recent work by Wang et al. 36 , EBV infected the full spectrum of the hematopoietic system including both lymphoid and myeloid lineages, so as hematopoietic stem cells (HSCs) in the chronic active EBV patients.
Besides, the majority of PTLDs after allo-HSCT were donor-derived, while those following SOT always recipient derived37,38. This research also supported this conclusion as all our T-cell PTLD patients and most of the reviewed cases were donor originated. The cell of origin after transplantation may influence the treatment choice. Immunosuppression tapering (IST) was first considered for low-risk PTLD patients with acceptable physical conditions. IST allows recovery of the host’s immune system, promoting EBV-specific T-lymphocytes to proliferate and control the disease 39 . More aggressive treatment, including chemotherapy, radiotherapy, surgery, rituximab, and cellular immunotherapy, such as donor lymphocyte infusion, would be considered when patients showed no response to IST 19 .
As the low incidence and dismal outcomes of T-cell PTLD, a key question is how to diagnose and intervene without delay. In our series, failure to empiric rituximab therapy (n = 4) led to pathological examination to confirm the PTLD subtype; therefore, these patients took a longer time to final diagnosis (59.5 [range: 31–114] days vs. 34 [20–48] days) leading to a lower response to CHOP-based therapy (3/4) compared with the other two patients who performed biopsy when symptoms onset. We recommended that patients with probable PTLD should perform biopsy promptly, and for patients who did not respond to IST and/or rituximab-based therapy, the diagnosis of T-cell PTLD should be considered. CHOP-based chemotherapy may be the preferred option, given it provided higher CR rate (87.5% vs. 29.0%) for T-PTLD compared to the other therapies.
This study has some limitations, including its retrospective designed, small sample size and heterogeneity of the patients. As the incidence of T-PTLD is rather low, the systemic review and the present case series would provide new insights into future studies for T-cell PTLD.
In summary, T-cell PTLD displays distinct characteristics compared with B-cell PTLD, which characterized by less connections to EBV, later occurrence, and poorer prognosis. Research is necessary to investigate the risk factors, how to diagnosis timely and more effective treatment for T-cell PTLD.
Footnotes
Author Contributions
X.H. and Y.C. designed the study. C.J. and J.H. wrote the manuscript. C.J., J.H., J.S., T.Y., Y.Z., M.H. H.Y., J.S., L.W., F.C., Y.C. and X.H. were involved in collecting, analyzing, or interpreting research data and writing the manuscript. C.J. and J.H. analyzed research data.
Ethical Approval
Institutional databases were retrospectively reviewed to extract demographic, clinical and genetic data. All procedures complied with the tenets of the Helsinki Declaration, and approved by Institutional Review Boards of participating centers. The requirement for written informed consent was waived, owing to the non-interventional and retrospective nature of the study.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (grant no. 82170206) and Shanghai Municipal Health Commission Project of Disciplines of Excellence (grant no. 20234Z0002).