
Abstract
While exagamglogene autotemcel (Casgevy) and lovotibeglogene autotemcel (Lyfgenia) have been approved by the US Food and Drug Administration (FDA) as the first cell-based gene therapies for the treatment of patients 12 years of age and older with sickle cell disease (SCD), this treatment is not universally accessible. Allogeneic hematopoietic stem cell transplant (HSCT) has the potential to eradicate the symptoms of patients with SCD, but a significant obstacle in HSCT for SCD is the availability of suitable donors, particularly human leukocyte antigen (HLA)-matched related donors. Furthermore, individuals with SCD face an elevated risk of complications during stem cell transplantation due to SCD-related tissue damage, endothelial activation, and inflammation. Therefore, it is imperative to consider optimal conditioning regimens and investigate HSCT from alternative donors. This review encompasses information on the use of HSCT in patients with SCD, including the indications for HSCT, conditioning regimens, alternative donors, and posttransplant outcomes.
Keywords
Introduction
Sickle hemoglobin (Hb) or HbS is the most common variant of Hb. A single nucleotide substitution (GTG for GAG) in the sixth codon of the β-globin gene results in the substitution of valine for glutamic acid on the surface of the variant β-globin (βS) chain (β6 Glu → Val)
1
. This minor structural change results in a profound change in the stability and solubility of the Hb molecule, allowing HbS to polymerize when deoxygenated since valine can dock with complementary sites on adjacent globin chains. Long polymers distort the red cell into a holly leaf, a crescent, or sickle shape, the so-called sickle cell, which is characterized by reduced deformability and increased adhesion to endothelial cells leading to vasculopathy and occlusion of capillary blood flow. The extent of polymerization depends on intraerythrocytic HbS concentration, degree of cell deoxygenation, pH, and the intracellular concentration of fetal hemoglobin (HbF)
2
. Symptoms of sickle cell disease (SCD) are related to anemia, vaso-occlusion by sickle-shaped cells, vasculopathy, and vasoconstriction from decreased nitric oxide availability, which lead to hypoxia of the tissues and painful vaso-occlusive episodes (VOE). More than 95% of newborns with SCD in developed countries survive into adulthood due to significant advances in supportive and preventive care for SCD in the last decade
3
. However, available drug treatments for patients with SCD are limited. Hydroxyurea (HU) has been the only US Food and Drug Administration (FDA)-approved drug for SCD until recently, leading to a significant effect on reducing pain episodes, acute chest syndrome, and transfusions, with minimal genotoxicity or carcinogenicity despite long-term use
4
. A better understanding of the disease pathophysiology has allowed progress on more focused pharmacological therapies and the development of new treatment modalities, including
Nevertheless, none of the pharmacologic treatments have been successful in complete suppression of SCD symptoms. Furthermore, advances in cellular and molecular biology have led to consider hematopoietic stem cell transplant (HSCT) or gene therapy with transduced autologous hematopoietic stem cells as optimal curative options in SCD. These cell treatments can establish complete or partial normal erythropoiesis4,5. In December 2023, the FDA approved exagamglogene autotemcel (Casgevy) and lovotibeglogene autotemcel (Lyfgenia), representing the first cell-based gene therapies for the treatment of patients 12 years of age and older with SCD and a history of recurrent vaso-occlusive crises (VOCs). Casgevy is the first FDA-approved therapy utilizing CRISPR/Cas9, a type of genome-editing technology and Lyfgenia uses a lentiviral vector (LVV; gene delivery vehicle) for genetic modification. Fig. 1 shows FDA-approved drugs and gene products for the management of SCD. Allogeneic HSCT can eliminate manifestations of patients with SCD but is limited by the absence of a full-matched donors and immune complications 6 . This review covers the use of HSCT in patients with SCD, including the indications for HSCT, conditioning regimens, alternative donors, and posttransplant outcomes.
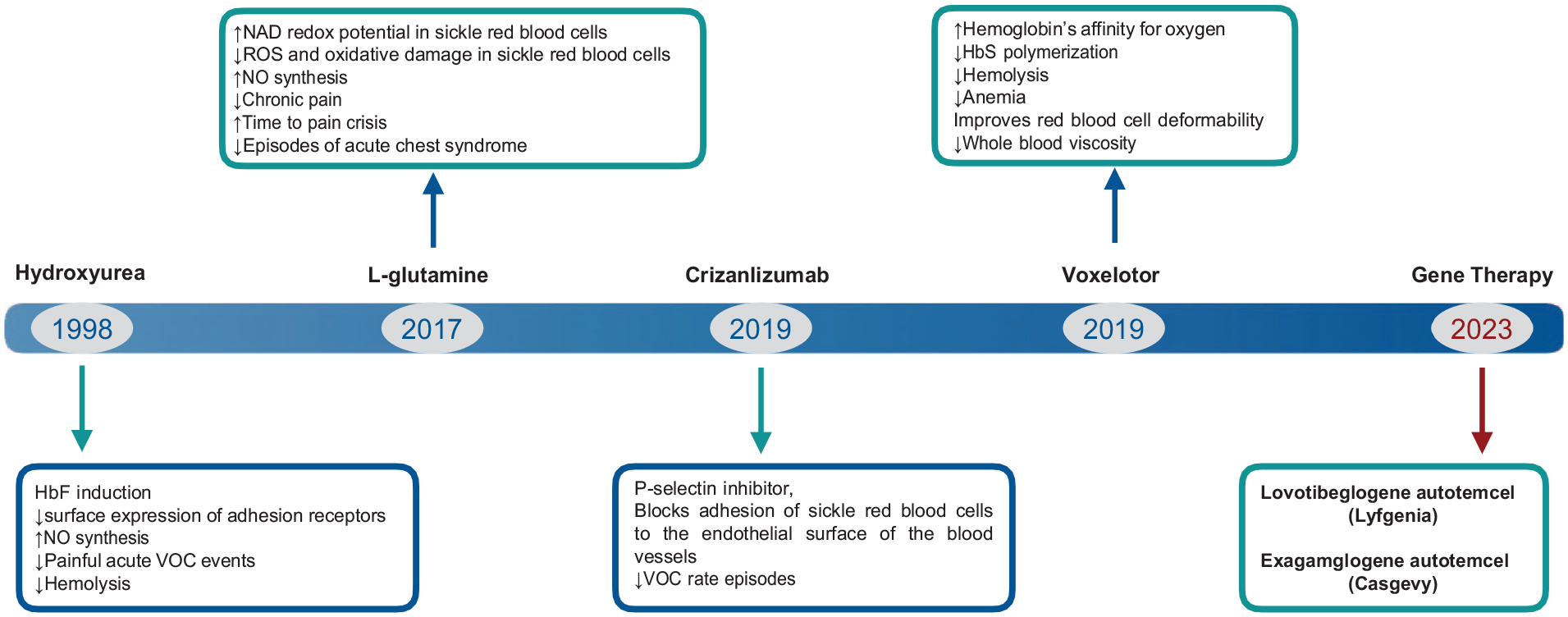
FDA-approved drugs and gene products for the management of SCD.
Allogeneic HSCT in SCD
HSCT is currently a curative treatment for severe SCD7–10. Five-year overall survival (OS) and event-free survival (EFS) of patients who received allo-HSCT from a human leukocyte antigen (HLA)-identical sibling donor have been reported to be 92%–94% and 84%–92%, respectively, with an improved OS in children younger than 16 years (95% vs 81%, P < 0.001) 11 . Allogeneic HSCT is offered most commonly to patients with serious SCD-related complications including stroke, recurrent VOCs, episodes of acute chest syndrome, and other significant organ damages12–14. Table 1 outlines the current indications for HSCT based on specific SCD complications and also the type of donor that might be considered [ie, matched related donor (MRD), haploidentical related donor, or matched unrelated donor (MUD)].
Current Indications for HSCT in Patients With SCD (One or More of the Following Complications) 15 .

HU: hydroxyurea; MRI/MRA: magnetic resonance imaging/ magnetic resonance angiography; MSD: matched sibling donor; MUD: matched unrelated donor; TCD: transcranial Doppler.
Given that there are no randomized clinical trials comparing stem cell transplant with conservative approaches in patients with SCD, a multidisciplinary guideline panel formed by the American Society of Hematology (ASH) addressed eight recommendations with very low certainty in the evidence, focused predominantly on which patients should be considered for HSCT (Table 2) 16 .
Summary of American Society of Hematology (ASH) Recommendations (R) for HSCT in Patients With SCD.

ACS: acute chest syndrome; HU: hydroxyurea; MSD: matched sibling donor; RIC: reduced intensity conditioning.
In an international survey conducted by the European Society for Blood and Marrow Transplantation (EBMT), Eurocord, and the Center for International Blood and Marrow Transplant Research (CIBMTR), including 1,000 patients with SCD who received HLA-identical sibling transplants mostly (87%) with a myeloablative conditioning (MAC) regimen and bone marrow as stem cell source (84%), the 5-year EFS and OS were 91.4% [95% confidence interval (CI), 89.6%–93.3%] and 92.9% (95% CI, 91.1%–94.6%), respectively. Noteworthily, by every 1-year increment in the age of recipients at HSCT, hazard ratio (HR) for acute graft versus host disease (GvHD) and treatment failure (graft failure or death) increased 4% and 10%, respectively. In patients 16 years and older at transplantation, a 2% increase in the HR for chronic GvHD was reported 17 . These results highlight the consensus document from the EBMT Inborn Errors Working Party and the Paediatric Diseases Working Party that recommends HSCT should be considered for symptomatic young patients who have an HLA-matched sibling donor (MSD) as early as possible before complications occur 18 . HSCT complications can exacerbate SCD-related organ damage, emphasizing the importance of careful recipient and donor selection, appropriate conditioning intensity, and effective GvHD prophylaxis. By addressing these factors, the aim is to minimize morbidity and mortality associated with HSCT and improve patient outcomes15,19,20.
MAC Regimens
Successful myeloablative transplantation trials in children with SCD from HLA-identical sibling donors have been reported in several studies. In a retrospective study conducted by Grupo Español de Trasplante de Médula Ósea en Niños (GETMON) and Grupo Español de Trasplante Hematopoyético (GETH), 45 children (from birth to 18 years of age) diagnosed of SCD received HSCT from an MSD. The majority of patients received a conditioning regimen based on busulfan and cyclophosphamide. The study found that the 3-year EFS and OS were 89.4% (95% CI, 73.9%–95.9%) and 92.1% (95% CI, 77.2%–97.4%), respectively. Notably, patients aged ≤5 years had 100% EFS and OS at 3 years posttransplant 21 .
Similarly, Cappelli et al. reported the outcomes of 736 patients with SCD who underwent HSCT from an HLA-identical sibling, in three different groups according to the age at transplant: 0–5 years: group 1, 6–15 years: group 2, and more than 15 years: group 3. Most of them received an MAC regimen. The 4-year OS was 100%, 95%, and 88% for groups 1, 2, and 3, respectively (P < 0.001). The 4-year EFS was 93% in group 1, 89% in group 2, and 81% in group 3 (P = 0.003). The overall cumulative incidence of grade II–IV acute GvHD was 10%, 18%, and 17% in groups 1, 2, and 3, respectively (P = 0.047). Chronic GvHD occurred in 9% of patients in group 1, 11% in group 2, and 20% in group 3 (P = 0.007) 22 .
Modification to traditional MAC regimens composed of busulfan in combination with highly immunosuppressive doses of cyclophosphamide and adding anti-thymocyte globulin (ATG) to decrease the risk of graft rejection have improved the outcome of allo-HSCT in pediatric patients with SCD. However, adult patients may experience excessive toxicity from myeloablative preparative regimens due to accumulated end-organ damage 23 . Table 3 shows a summary of several studies using MAC regimens in patients with SCD.
Myeloablative Conditioning Regimen in Patients With SCD.
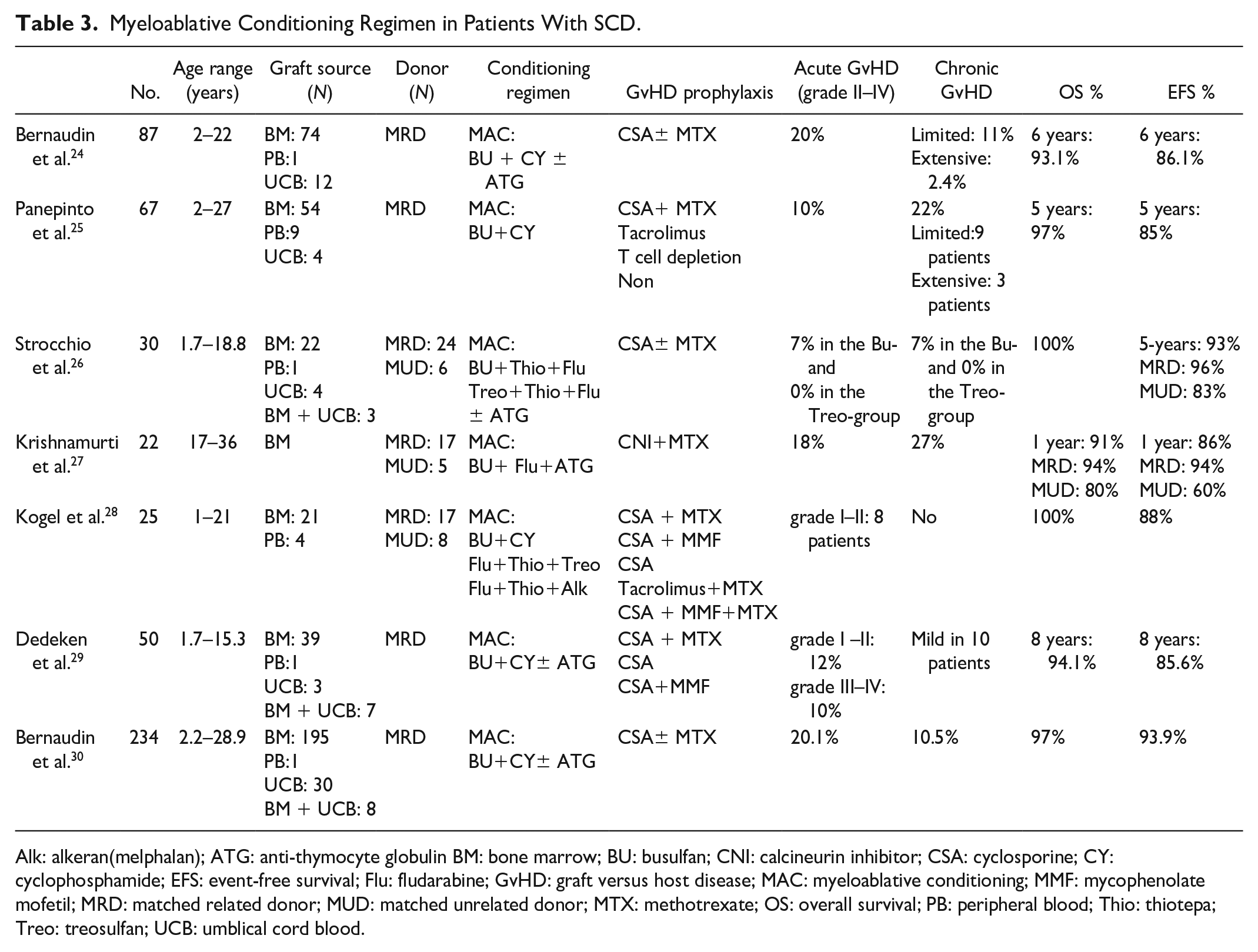
Alk: alkeran(melphalan); ATG: anti-thymocyte globulin BM: bone marrow; BU: busulfan; CNI: calcineurin inhibitor; CSA: cyclosporine; CY: cyclophosphamide; EFS: event-free survival; Flu: fludarabine; GvHD: graft versus host disease; MAC: myeloablative conditioning; MMF: mycophenolate mofetil; MRD: matched related donor; MUD: matched unrelated donor; MTX: methotrexate; OS: overall survival; PB: peripheral blood; Thio: thiotepa; Treo: treosulfan; UCB: umblical cord blood.
Reduced Intensity Conditioning Regimens
Adult patients with SCD are at high risk of experiencing complications when undergoing stem cell transplantation, due to SCD-mediated tissue damage, endothelial activation, and inflammation. These complications include drug toxicity, GvHD, and graft rejection 31 . Reduced intensity conditioning (RIC) regimens have been developed to decrease toxicities related to MAC regimens and make HSCT more acceptable and well-tolerable for patients with SCD. To reduce transplant-related toxicity of MAC regimens, the feasibility of a highly immunosuppressive conditioning (RIC) regimen consisting of fludarabine, melphalan, and either thiotepa or total lymphoid irradiation (TLI) was explored in eight children with SCD. After a median follow-up of 4 years, three patients had mixed lymphocyte donor chimerism, and all patients had 100% donor erythropoiesis 32 .
Krishnamurti et al. enrolled seven pediatric patients with SCD in a study to evaluate the safety and efficacy of HSCT from an HLA-MSD following an RIC regimen consisting of busulfan, fludarabine, equine ATG, and TLI. Despite mixed chimerism in the mononuclear and lymphoid cells, complete resolution of SCD-related symptoms was observed in the six engrafted patients, and erythropoiesis of complete or predominantly donor origin was detected by red blood cell–specific chimerism assays 33 . These findings demonstrate the curative potential of allogeneic hematopoietic cell transplantation (HCT) after an RIC regimen in patients with SCD.
Nonmyeloablative Conditioning Regimens
Stable mixed chimerism, defined as a mixture of the recipient- and donor-derived lymphohematopoiesis, has been associated with improved laboratory parameters and the prevention of SCD-related complications. Studies have shown that at least 20% myeloid donor chimerism is necessary to reverse the SCD phenotype. In this regard, nonmyeloablative (NMA) conditioning regimens with lower rates of GvHD and HSCT-related toxicity are found of particular importance to apply in severely affected adults 34 .
Saraf et al. presented the outcomes of stem cell transplantation in 13 high-risk adult patients with SCD using an NMA preparative regimen including alemtuzumab and total body irradiation (TBI) of 300 cGy. A stable mixed donor/recipient chimerism was maintained in 12 patients (92%), which was associated with normalized Hb concentrations and also decreasing veno-occlusive crisis. None of the recipients experienced acute or chronic GvHD, and there was 100% OS at a median follow-up of 22 months 35 .
In another study, 17 adult patients with SCD with a median age of 23.5 (14–39) years were conditioned with an NMA conditioning regimen including alemtuzumab, TBI, and transplanted from fully MSDs. All donors were fully matched siblings. After the transplant, none of the patients developed acute or chronic GvHD, and also no SCD-related events developed. All patients had full donor myeloid engraftment and normalization of blood count. At the last follow-up, OS and EFS were 100% and 88.9%, respectively. These findings indicate that NMA preparative regimen is safe, feasible, and effective in reducing SCD-associated complications 36 .
Table 4 summarizes several studies using reduced intensity and NMA conditioning regimens in patients with SCD.
Nonmyeloablative/RIC Conditioning Regimen in Patients With SCD.
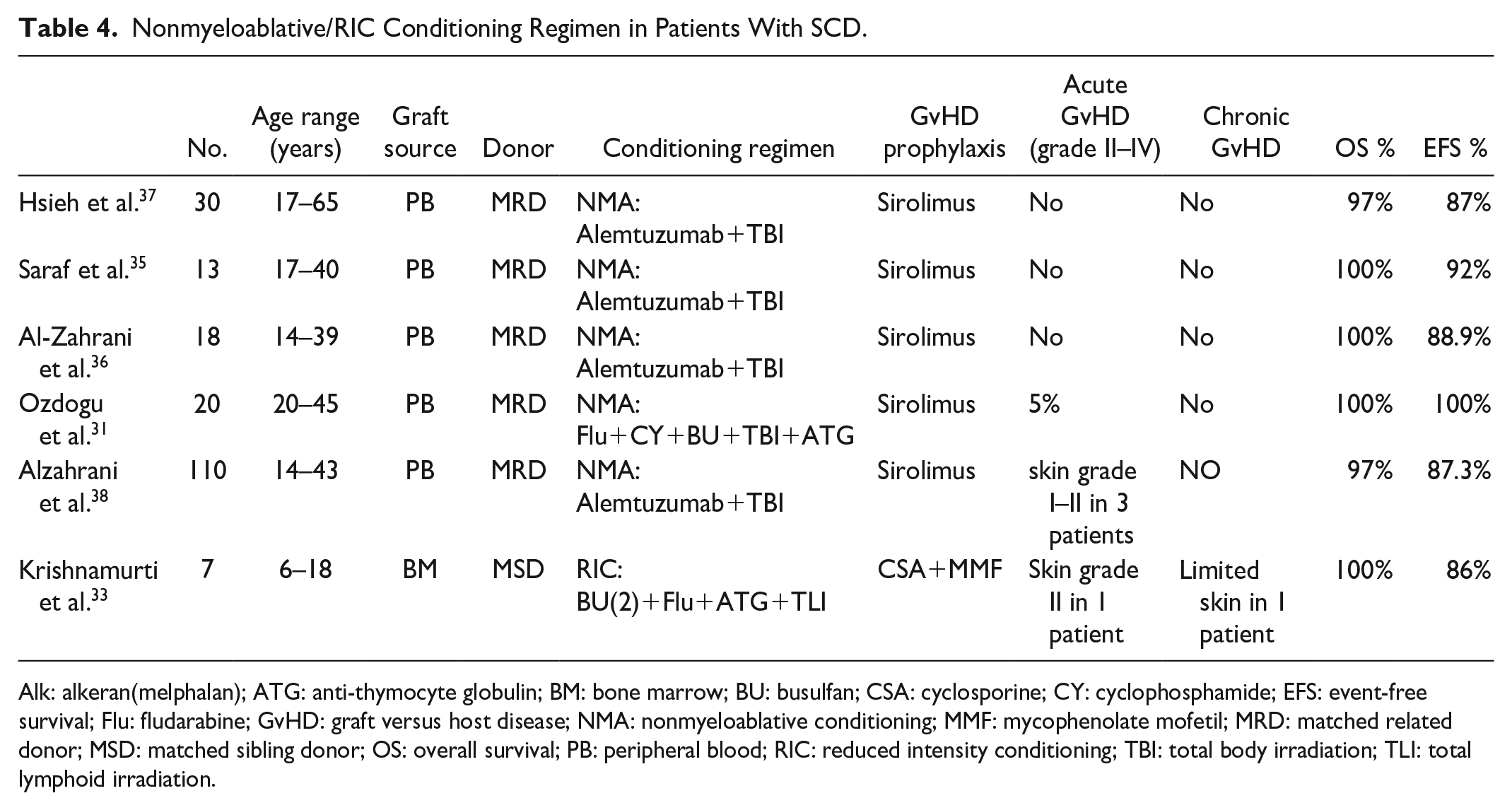
Alk: alkeran(melphalan); ATG: anti-thymocyte globulin; BM: bone marrow; BU: busulfan; CSA: cyclosporine; CY: cyclophosphamide; EFS: event-free survival; Flu: fludarabine; GvHD: graft versus host disease; NMA: nonmyeloablative conditioning; MMF: mycophenolate mofetil; MRD: matched related donor; MSD: matched sibling donor; OS: overall survival; PB: peripheral blood; RIC: reduced intensity conditioning; TBI: total body irradiation; TLI: total lymphoid irradiation.
Alternative Donors
Although HSCT from HLA-MRDs has been encouraging in patients with SCD, a major proportion of these patients do not have a suitable HLA-identical sibling donor. Alternative stem cell sources including matched or mismatched unrelated donors, umbilical cord blood (UCB), or haploidentical related donors have expanded the possibility of finding suitable donors for patients with SCD who meet the criteria for HSCT11,39. However, the likelihood of finding a potentially MUD is dependent on the ethnic and racial background, with the highest probability among white European people and the lowest in black people of South or Central American descent40–42.
Haploidentical HSCT in patients with SCD has been associated with encouraging results. An NMA regimen including fludarabine, cyclophosphamide, and TBI was used in 14 patients with SCD who underwent haploidentical stem cell transplantation. GvHD prophylaxis consisted of high-dose posttransplantation cyclophosphamide (PTCy), mycophenolate mofetil, and tacrolimus or sirolimus. Although none of the patients developed GvHD, conditioning regimens were well tolerated and no transplant-related mortality was observed; only 57% of patients achieved stable engraftment 43 .
A phase I/II trial was conducted by Fitzhugh et al., using alemtuzumab, 400 cGy TBI, and escalating doses of PTCy (cumulative cyclophosphamide dose, group 1: no PTCy; group 2: 50 mg/kg body weight and group 3: 100 mg/kg body weight) in 21 patients with SCD who underwent haploidentical HSCT. Although the engraftment rate at day +100 improved in group 3 compared with the other two groups (83% vs 63% in group 2 and 33% in group 1), no patient achieved complete 100% donor chimerism. The mean percentage of donor myeloid chimerism in cohort 3 patients was 52.1% at 3 months, 84.8% at 6 months, and 52% at 3 years posttransplant 13 .
In a prospective phase 2 clinical trial, 19 children and adolescent patients with SCD proceeded to CD34+ enriched haploidentical HSCT following a myeloimmunoablative preparative regimen. Patients received HU and azathioprine from day −59 to day −11 and were then conditioned with fludarabine, busulfan, thiotepa, cyclophosphamide, TBI, and ATG. Mononuclear cells cryopreserved as a source of MNC add-back (T cells). The cumulative incidence of grades II–IV acute GvHD was 6.2% and moderate and/or severe chronic GvHD was 6.7%. Peripheral blood and red blood cell mean donor chimerism at 1 year after transplantation was 97.1% and 96.4%, respectively. The 2-year probability of EFS and OS was 84%; however, none of the patients demonstrated SCD symptoms 44 .
Table 5 summarizes several studies using haploidentical donors in patients with SCD.
Haploidentical Hematopoietic Stem Cell Transplantation in Patients With SCD.
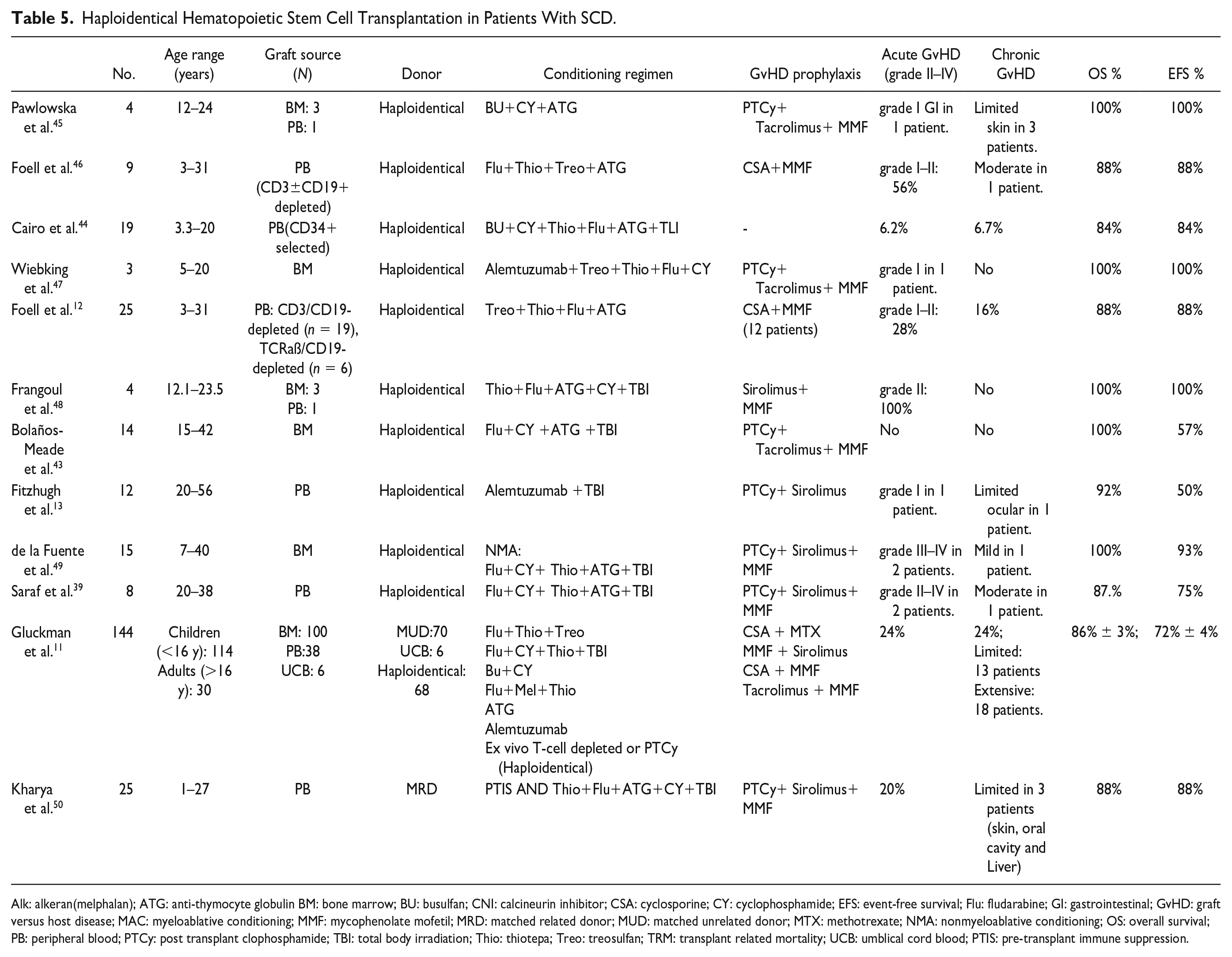
Alk: alkeran(melphalan); ATG: anti-thymocyte globulin BM: bone marrow; BU: busulfan; CNI: calcineurin inhibitor; CSA: cyclosporine; CY: cyclophosphamide; EFS: event-free survival; Flu: fludarabine; GI: gastrointestinal; GvHD: graft versus host disease; MAC: myeloablative conditioning; MMF: mycophenolate mofetil; MRD: matched related donor; MUD: matched unrelated donor; MTX: methotrexate; NMA: nonmyeloablative conditioning; OS: overall survival; PB: peripheral blood; PTCy: post transplant clophosphamide; TBI: total body irradiation; Thio: thiotepa; Treo: treosulfan; TRM: transplant related mortality; UCB: umblical cord blood; PTIS: pre-transplant immune suppression.
Posttransplant Outcomes
In a study by Lucarelli et al., 40 consecutive patients with SCD (13 patients with the nonblack African variant and 27 with the black African variant) who received MAC regimen HSCT from HLA-identical sibling donors were enrolled. None of the surviving patients experienced sickle cell anemia (SCA)-related events after transplantation such as pain, stroke, or acute chest syndrome 51 .
Eleven consecutive children with SCD underwent HSCT with a treosulfan, thiotepa, fludarabine, and ATG conditioning regimen from MSD (n = 7), haploidentical parent (n = 2), MUD (n = 1), or a mismatched unrelated donor (n = 1). Full donor chimerism was achieved in six patients and stable mixed chimerism was observed in five patients (45%). No episodes of acute chest syndrome, stroke, or other SCD manifestations occurred. Cerebral vasculopathy improved in three of the five evaluable patients 52 .
Among 26 patients with stable donor engraftment following HSCT from MSD bone marrow allografts, complications related to SCD resolved in 22 recipients without further episodes of pain, stroke, or acute chest syndrome. For patients who had a prior history of stroke, cerebral magnetic resonance imaging (MRI) results were stable or improved after transplant. Eight patients were transplanted due to recurrent acute chest syndrome, of whom seven patients had stable pulmonary function after HSCT 53 .
A single-center, retrospective study enrolled 20 consecutive patients who underwent HSCT from HLA-MRDs with a conditioning regimen of fludarabine, busulfan, cyclophosphamide, ATG, and TBI. Although 90% of recipients had a prior history of two or more painful crises before transplant, none of them experienced painful episodes, after transplant with a significant decrease in hospitalization and narcotic use 31 .
Cerebral vasculopathy was the main indication for HSCT in 55 patients with SCD ranging from 2 to 22 years of age in a study by Bernaudin F et al. All patients received transplantation from a sibling donor after an MAC regimen. Thirty-six patients had a pretransplantation history of stroke, of whom only two had a recurrence of stroke; one experienced a transient ischemic attack (TIA) on day +10, and the other with Moyamoya disease had a fatal intracranial hemorrhage on day 32 after transplantation. Vascular stenosis resolved in 5 patients, remained stable in 16 patients, and progressed in 2 patients. Cerebral velocities were significantly reduced 1 year after transplantation in the 49 assessable patients and normalized in 2 patients. However, no new silent ischemic lesions were detected in patients with successful engraftment 24 .
Seventeen patients proceeded to HSCT with an NMA bone marrow transplantation, from HLA-MSDs (N = 14) and HLA-haploidentical related donors (N = 3) with a median age of 30 years (range, 15–46). Evidence of long-term engraftment was achieved in 11 patients. No clinical evidence of new cerebrovascular events, acute chest syndrome, or priapism has been reported in engrafted patients 43 .
The National Institutes of Health (NIH) administered an NMA conditioning regimen including alemtuzumab and 300 cGy TBI to 16 children and adolescents with SCD prior to HSCT. All patients achieved stable mixed donor chimerism that was sufficient to maintain HbS levels below 50%. So no sickling crises occurred after transplant 54 .
In the setting of allogeneic NMA stem cell transplant, 67 patients with SCD were followed up by monitoring of myeloid and lymphoid chimerism, Hb and HbS levels, and SCD symptoms every 6–12 months. Three of these patients experienced mixed donor myeloid chimerism. However, as long as the mean mixed donor myeloid chimerism was more than 20%, mean percentage of HbS remained below 50% which was sufficient to reverse SCD-related symptoms 13 .
A retrospective study of 95 patients with SCD who underwent related or unrelated donor HSCT with RIC regimen in 66% of cases concluded that stable donor chimerism greater than 25% is associated with resolution of SCD-related symptoms 55 .
Twenty-two pediatric patients with SCD underwent HSCT at St. Jude Children’s Research Hospital, from sibling MRD (N = 14) or haploidentical donor (N = 8) with MAC and RIC regimens, respectively. After 5 years posttransplant, none of the engrafted patients experienced clinical evidence of cerebrovascular accident (CVA). Imaging studies by MRI and magnetic resonance angiography (MRA) revealed improvement in white matter changes and stable or even improved vessel abnormalities. Cerebral velocity also decreased significantly in transcranial Doppler (TCD) ultrasound studies. To evaluate the risk of osteonecrosis, all patients underwent imaging studies (MRI or radiography). Three patients with evidence of avascular necrosis (AVN) pre-HSCT had stable findings by 5 years after transplant. However, two patients developed AVN 2 years after HSCT. The mean creatinine clearance decreased from 158 ± 55.7 to 103.5 ± 24 ml/min/1.73 m2 in the MRD cohort and from 98 ± 33 to 91 ± 47 ml/min/1.73 m2 in the haploidentical cohort. At the last follow-up, all patients had normal urinalysis results, with no proteinuria or hematuria 40 .
Eighteen patients with a median age of 8.9 years (range, 2.3–20.2) underwent an HLA-matched sibling allo-HSCT, of whom eight patients received allografts from sickle trait donors. One and 2 years after transplant, they had a median HbS of 38% and 39%, respectively. Using tricuspid regurgitation (TR) jet velocity on transthoracic echocardiogram to assess pulmonary hypertension (PH), no demonstrable evidence of PH was detected 1 and 2 years after transplant even in patients with mild to moderate PH at baseline. Pulmonary function tests (PFTs) performed pre-allo-HSCT demonstrated restrictive lung disease in three patients and obstructive lung disease in one patient. However, PFT was stable or improved 2 years after HSCT 56 .
In an NMA transplant study, 30 patients aged 16–65 years with severe SCD enrolled. At the median follow-up of 3.6 years, the mean donor myeloid cell chimerism was 86% (95% CI, 70%–100%). In nine patients with a prior history of stroke or abnormal central nervous system vessels, post-HSCT brain MRI and angiography remained unchanged in patients with sustained engraftment. In patients with TR velocity higher than 2.5 m/s, their mean velocity decreased to 2.57 m/s (95% CI, 2.44–2.69) 1 month, to 2.43 m/s (95% CI, 2.12–2.70) 1 year, and to 2.33 m/s (95% CI, 2.14–2.51) 3 years after HSCT 37 .
To evaluate the safety and efficacy of HSCT following an RIC regimen, seven patients with high-risk SCD received bone marrow transplants from an HLA-MSD. Six patients engrafted, of whom two demonstrated full donor chimerism and four had mixed donor chimerism at levels ranging from 71% to 95%. All four patients with partial donor chimerism demonstrated >90% donor red blood cell (RBC) chimerism and all engrafted patients revealed the percentage of peripheral blood HbS from the donor’s genotype. One year after HSCT, all seven patients had stable brain MRI findings and PFTs. In five engrafted patients without prior history of splenectomy, splenic regeneration was found on a spleen scan 33 .
Thirteen adult patients with high-risk SCD underwent HSCT using a chemotherapy-free, alemtuzumab/TBI 300 cGy regimen. Stable mixed donor chimerism was achieved in 12 patients (92%) and no acute or chronic GvHD occurred. After transplant, only one engrafted patient with a prior history of recurrent VOCs required readmission to the hospital. Cardiac and pulmonary functions improved at 1 year 35 .
A multicenter pilot study was conducted to investigate the safety and feasibility of bone marrow transplantation in 22 adult patients with severe SCD using a reduced toxicity preparative regimen. Although mixed donor chimerism in the lymphoid cells was observed in 21 patients, all patients achieved full donor myeloid chimerism at all time points after transplant. Of 17 patients with a baseline brain MRI, all had a stable MRI at 1 year after HSCT. No significant changes occurred in PFT parameters including forced expiratory volume in 1 s (FEV1), FEV1/FVC (forced vital capacity), and total lung capacity (TLC). Considering TRJ velocity, it was ≥2.7 m/s in five patients, of whom three patients did not have TRJ recorded in follow-up studies, and in the two other patients, TRJ decreased to less than 2.7 m/s. In one patient with TRJ of 2.4 m/s before transplant, it increased at 1-year post-BMT 27 .
Fifty-six patients with SCD underwent NMA HLA-matched sibling or haploidentical donor HSCT accompanied by successful engraftment and reversal of the SCD phenotype in 44 patients (79%). After transplant, the estimated TRJ velocity remained stable at any time point. In 25 patients with a TR velocity ≥2.5 m/s at baseline, velocity decreased significantly from a median of 2.7–2.3 m/s at 1 year (P < 0.005). Serum creatinine increased from 1.0 ± 1.5 mg/dl at the time of transplantation to 1.4 ± 2.8 mg/dl (P ≤ 0.005) 1 year after HSCT 57 .
Fifty patients affected by SCD underwent HSCT from HLA-genoidentical siblings in 49 patients and HLA-phenoidentical father in one patient. The source of stem cells was bone marrow in 48 patients and UCB in two. Engraftment occurred in 47 patients and 2 patients experienced rejection. Mixed chimerism was present in six patients. The probability of OS and EFS was 93% and 82%, respectively. No recurrence of strokes was reported among the four patients who had a prior history of stroke before transplantation. Osteonecrosis of hips and shoulders was present in three patients at the time of transplantation with no improvement 3 years after HSCT 58 .
Table 6 summarizes the post-HSCT neurological outcomes in patients with SCD.
Summary of Post-HSCT Neurological Outcomes in Patients With SCD.

Genetically Manipulated Autologous HSCT in SCD
Human globin is composed of four subunits, encoded by a genomic structure comprising three exons and two introns 64 . The α- and the β-globin loci are located in chromosomes 16 and 11, respectively. In SCD, the βS mutation which is a single mutation in the sixth codon of the first exon in chromosome 11 leads to the replacement of adenine nitrogen base by thymine (HBB; 11p15.4, β6 GAG-GTG), changing a glutamic acid into a valine and formation of the HbS tetramer (α2β2S)65,66. In gene therapy as a potential cure for SCD, the goal is to permanently correct the HBB mutation in the patient’s autologous pluripotent stem cells with the ability to expand and differentiate into normal erythrocytes and highly expressing the normal HBB gene 67 . In the past decades, several endeavors have been directed toward efficiently delivering a functional copy of the HBB gene into human hematopoietic stem/progenitor cells (HSPCs), with the ability of homing in patient’s marrow environment, differentiating to erythrocytes68,69. Several approaches are being investigated to correct the genetic defect in HSPC of patients with SCD.
The efficient introduction of a fully functional β-like globin transgene into HSPCs is primarily indebted of the HIV-1-derived LVV emergence 70 . Moving forward, the discovery of the locus control regions (LCRs) enabled boosting the expression of β-like globin 71 . The introduction of critical LCR enhancers into the LVV and the optimization of the vector titer have made the rescue of SCD models possible 72 . Moreover, to optimize gene addition strategies, the expression of a potent anti-sickling globin transgene, inhibiting HbS polymerization, enhancement of vector infectivity, and HSPC transduction are being explored. Considering that the elevated levels of fetal γ-globin could amend the clinical course of SCD by inhibiting HbS polymerization, the therapeutic strategies also aimed at forcing the β- to γ-globin switching in concurrence with the reduction of sickle β-globin gene expression 73 .
In genome editing, the DNA double-strand breaks (DSBs) induced at selected genomic loci are repaired by means of homology-directed repair (HDR) using a homologous DNA used as the repair template. To correct the SCD mutation via HDR in HSPCs, clustered regularly interspaced short palindromic repeat (CRISPR)-associated Cas9 nucleases or zinc finger nucleases (ZFNs) together with single-stranded oligonucleotide donors or integrase-defective LVVs carrying the donor templates are used74–77. Genome correction of the SCD point mutation in exon 1 has been achieved by validated nucleases targeting specific locations of various specific HBB mutations. The ease and robustness of the CRISPR/Cas9 system has become the preferred choice in recent years for making a specific DSB in the HBB locus and achieving HDR to correct a specific HBB gene mutation78–80. On December 8, 2023, the FDA approved two autologous gene therapy products, Lyfgenia which utilizes an LVV-based strategy to transduce autologous HSCs with an anti-sickling globin and Casgevy which is the first ever FDA-approved CRISPR-based gene therapy product, utilizing CRISPR-Cas9 editing at the erythroid-specific enhancer of the BCL11A gene to enhance fetal Hb production 81 .
Conclusion
Patients with SCD face challenges with HSCT, which is currently an established curative intervention. These challenges include donor availability and morbidity related to age and disease severity. To improve patient outcomes, it is crucial to consider optimal conditioning regimens, careful recipient, and donor selection, and exploring HSCT from alternative donors. Ongoing research is needed to further improve the safety and efficacy of HSCT procedures and optimal preparative conditioning regimens.
Footnotes
Author Contributions
T.R. and A. Ki contributed to the design and conceived of the program, directed the project, reviewed the studies, wrote, and edited the manuscript. H.A. assessed the studies, extracted the data, wrote part of the manuscript, edited, and critically revised the manuscript. S.R., M.R.R., S.A.M. (Seied Amirhossein Mirhosseini), and N.K. assessed the studies, extracted the data, and wrote part of the manuscript. A. Ka wrote part of the manuscript and directed the project. T.R., G.J., S.A.M. (Seied Asadollah Mousavi), and A. Ka critically revised the manuscript. All authors read and approved the final manuscript and accept accountabilities for all aspects of this work.
Availability of Data and Materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during this study.
Ethical Approval
This study was approved by our institutional review board.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.