
Abstract
Microglia are associated with a wide range of both neuroprotective and neuroinflammatory functions in the central nervous system (CNS) during development and throughout lifespan. Chronically activated and dysfunctional microglia are found in many diseases and disorders, such as Alzheimer’s disease, Parkinson’s disease, and CNS-related injuries, and can accelerate or worsen the condition. Transplantation studies designed to replace and supplement dysfunctional microglia with healthy microglia offer a promising strategy for addressing microglia-mediated neuroinflammation and pathologies. This review will cover microglial involvement in neurological diseases and disorders and CNS-related injuries, current microglial transplantation strategies, and different approaches and considerations for generating exogenic microglia.
Keywords
Introduction
The maintenance of tissue homeostasis in the central nervous system (CNS) during development and life is largely due to the presence of healthy and functioning microglia; however, after neuronal injury or insult, microglia can enhance either cytotoxic or protective factors to assist in tissue repair. Sustained activation of microglia is damaging, and the feedback from neighboring cells can exacerbate the chronic inflammation and lead to cognitive decline and many neurodegenerative diseases and disorders. Methods for resolving microglia-mediated neuroinflammation include novel treatment strategies like transplantation of exogenic microglia. Exogenic microglia have the potential to protect against extensive inflammation and neuronal damage after the initial injury and promote repair.
In this review, we will cover what is known about microglial involvement in different neurodegenerative diseases and disorders, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and CNS-related injuries such as stroke, traumatic brain injury (TBI), and spinal cord injury (SCI; Fig. 1). We will describe commonly used cell types for transplantation, current microglial transplantation methods, and other potential approaches for generating exogenic microglia and transplanting them toward beneficial outcomes.
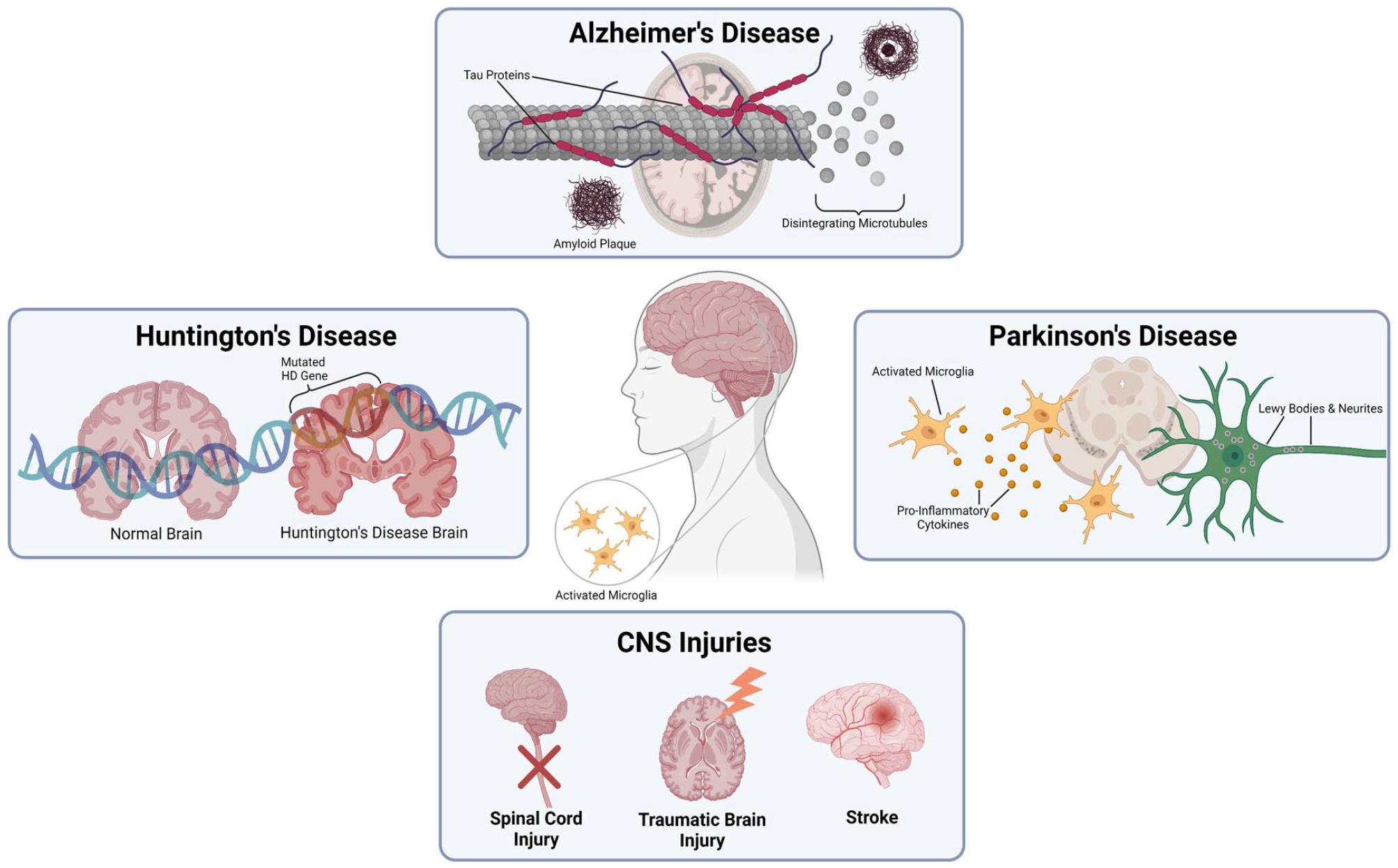
Diseases and disorders associated with microglial dysfunction. Disease pathology of many different neurodegenerative diseases and disorders, such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and CNS-related injuries, includes the chronic activation of dysfunctional microglia.
Function of Microglia
Origin and Function
Microglia are the resident immune cells of the CNS that compose approximately 7% of the nonneuronal population of cells in the brain and show a high degree of plasticity1–4. Microglia play key roles in shaping and maintaining the local environment by regulating synaptic formation, supporting neural plasticity, phagocytizing cells and debris, and releasing inflammatory modulators 5 . Their close interactions with neurons and other cells in the brain, such as astrocytes and oligodendrocytes, allow microglia to contribute to the development of many types of CNS-related injuries and neurodegenerative disease pathologies6–8.
Activation Profiles and Phenotypes
The ability for microglia to transform, depending on their role in maturation, proliferation, or response to damage and disease in the CNS, involves changes in gene expression, morphology, migration, and metabolism. Like macrophages, microglia can be categorized on a spectrum of functional activities and different states of polarity from resting (M0) to activated pro-inflammatory (M1) or alternatively activated anti-inflammatory (M2) due to environmental conditions. This switch in phenotype has been well characterized in many models of CNS injury and neurodegenerative diseases and disorders9–12. It is important to note that the M0/M1/M2 trichotomy is a vast oversimplification of the different activation states, as microglia can transform along a multidirectional spectrum of functionality of overlapping phenotypes. For example, M2 microglia can be further divided into subpopulations of M2a, M2b, and M2c13–15, all of which have unique physiological features and biological functions. More recently, nine different microglial states were identified based on gene expression, patterns, and activity. By turning specific genes on or off, microglia can shift from one state to the other 16 . Understanding how genetic variations may contribute to different diseases and disorders of the brain could provide insight on how to shift microglia from a pathogenic state to a healthy state.
Molecular Conversations From Development to Dysfunction
Microglia constantly monitor their microenvironment within the brain, contributing to the immunological defense of the CNS. As microglia surveil their surroundings, they must communicate with adjacent cells, integrate various signals, and respond in the appropriate manner. These microenvironments include neurons, astrocytes, and T-cells, which all engage in crosstalk with microglia 17 . Recent reports have indicated that neurons take an active role in sending “on” or “off” signals to direct microglia activation in their microenvironment18,19.
Interactions with neurons
Maintaining neuronal homeostasis requires continuous crosstalk between microglia and neurons. Microglia express a diversity of neurotransmitter receptors for sending neuronal activity. Stimulation of these receptors regulates activity such as cytokine production, phagocytosis, and cellular motility 20 . In a reciprocal and bidirectional relationship, neurons also express receptors that can be activated by molecules released by microglia, enabling microglia to influence neurotransmission necessary for housekeeping functions during development. An example of this includes the mutual regulation of the brain involving both neurons and microglia expressing ionotropic and metabotropic glutamatergic receptors (MLCuRs) to transform microglia into a neuroprotective or neurotoxic phenotype through the stimulation of MLCuR subtypes, such as group III MLCuRs and group II MLCuRs21–24. Purinergic signaling and adenosine signaling also play a key role in the dynamics of microglia and neuron communication, enabling control of microglial motility, migration, and function25–28.
Interactions with chemokines
Neurons can produce fractalkine, a chemokine that aids in cell chemotaxis and interacts with microglial receptor chemokine receptor 1 (CX3CR1), to signal endothelial adhesion and chemo-attraction29,30. Deficiency in chemokine ligand 1 (CX3CL1)/CX3CR1 signaling is correlated with neurodegeneration, as reflected by the neuronal loss in PD and amyotrophic lateral sclerosis (ALS) models as well as an accelerated disease progression in ALS and AD29,31,32.
Interactions with the complement system
Microglia serve as the effector cells in the complement system and determine which synapses should be eliminated in normal development, whereas reactivation of these complement components and microglia during adulthood result in neurodegeneration 33 . Membrane proteins and their receptors that make up the complement system are expressed in both neurons and microglia, playing a role in microglial activity by altering morphology, motility, and function in response to injury as well as fine-tuning brain circuitry during development34,35. Overactivation and overexpression of complement factors result in altered functions of microglia that contribute to an inflammatory milieu and may induce reactive astrocytes that are often seen in neurodegenerative disorders such as AD and PD20,36–39.
Interactions with astrocytes
In the developing brain, the molecular conversation between microglia and astrocytes begins as these cells populate the brain parenchyma, modulating brain functions and neuronal activity. This relationship has been implicated in the progression of diverse neuropathologies, and both cell types can induce activation and control cellular functions of the other40,41. As the primary cell to respond to insult to the CNS, microglia secrete molecular factors that determine both fate and function of astrocytes42–45. For example, microglia can induce astrocytes to switch to a neuroprotective phenotype through the downregulation of purinergic receptor P2RY1 46 , while interleukin (IL)-1a, tumor necrosis factor (TNF)-a, and complement component C1q stimulate a reactive inflammatory phenotype in astrocytes39,47. Through the release of a variety of molecules, including cytokines, chemokines, growth factors, and adenosine triphosphate (ATP), the molecular communication between microglia and astrocytes contributes to brain homeostasis, function, and development; whereas, miscommunication between the two cell types may result in neurological dysfunction 46 .
Role in Neuroinflammation
The involvement of microglia in several maintenance and regulatory processes in the CNS contributes to the growing body of evidence that microglia also mediate neuroinflammation that is associated with disease progression. Within the brain, microglia switch from M2 to a more inflammatory M1 state in response to both injury and aging48–51. The distribution of microglial polarity is highly dependent on the stage and severity of the disease48,50,51, as both the population and proportion of M1 and M2 microglia change depending on disease states12,52,53. Understanding the stage-specific switching of microglial phenotypes provides a therapeutic potential of microglia for cell transplantation therapy for treating neurodegenerative disorders, diseases, and injuries.
Role in Aging
The role of microglia in the maintenance of homeostasis contributes to remodeling of neural networks and includes the removal of aberrant proteins and debris that accumulate in the brain over time54,55. Aging involves physiological changes that are associated with oxidative stress, telomere shortening, DNA damage, decrease in synaptic plasticity, and chronic inflammation56–58. Microglia may contribute to this mild, chronic, age-associated inflammation by expressing pro-inflammatory cytokines, such as TNF-α, TNF-β, IL-1α, IL-1β, and IL-6 59 . The accumulation of aberrant proteins associated with neurodegenerative diseases, such as amyloid beta (Aβ), α synuclein, and apolipoprotein E4, may be due to the loss of phagocytic efficiency in microglia as a result of the decrease in their regulatory functions 60 . The senescence and dysfunction of microglia with age and neurodegenerative conditions suggest that microglia replacement by transplantation may be an approach for rejuvenating the microglia population within the brain and restore neurological function.
Microglial Transplantation
Sources of Microglia
A source of available cells is required for microglial transplantation to be a possible therapy for various neurological conditions. Sources of exogenic microglia range from donor animals to cell cultures, including fetal microglia derived from early days of development and induced pluripotent stem cell (iPSC)–derived microglia generated through ex vivo culture conditions or blastocyst complementation techniques61–63.
Donor microglia
Fetal, postnatal, and adult microglia are potential sources for exogenic microglia. Exogenic microglia are harvested and isolated from brain tissue of rodents and placed in in vitro cultures 62 . In vitro exogenic microglia can be used for transplantation and further identified and determined through selective polarization based on their anti-inflammatory or pro-inflammatory gene expression or cytokine release 64 .
Microglia extracted from animals of various ages and cultured in vitro express characteristics that change with age. For example, embryonic cultured microglia are much more ameboid shaped than their adult counterparts, which are typically more ramified 62 . In addition to morphological changes, alterations in microglial reactivity also occur. The reactivity of microglia can be determined by using endogenous microglial activators (eg, Aβ) to study the inflammatory response of microglia 65 . One study observed that neonatal microglia are more responsive to ATP through purinergic receptors, compared with adult microglia counterparts when cultured in vitro 62 . However, a more recent study has shown that cultured neonatal microglia from postnatal day 3 were less responsive to toll-like receptor (TLR) agonists compared with cultured adult microglia 66 . These microglial reactivity studies were tested in cultured microglia, which may alter the reactive state observed, and should be interpreted with caution. In addition, the age exogenic donor microglia are variable and can complicate the transplantation process in terms of engraftment and survivability. To match the appropriate species, human microglia would need to be utilized for transplantation, which has profound ethical implications.
IPSC-derived microglia
Microglia derived from iPSCs provide an accessible and renewable source of exogenic microglia for treating a variety of neurological disorders that result from microglial dysfunction. Generating microglia from iPSCs is challenging, due to their origin from the yolk sac erythromyeloid progenitors 67 . Human embryonic stem cells (ESCs) are one of the first cells used to derive a heterogeneous population of macrophage progenitor colonies. These protocols were refined to produce a pure macrophage population 68 . Studies have demonstrated that iPSC-derived macrophages are MYB-independent yolk sac–derived embryonic macrophages, which make them ideal microglial progenitors. iPSC-derived macrophages co-cultured with neurons behave like microglia with typical morphology, movement, transcriptomes, and cytokine release69,70. Several groups have developed protocols to produce ESC- and iPSC-derived microglia rather than macrophages and have generated microglia or microglia-like cells (MLCs) that recapitulate characteristics and behaviors of in vivo microglia63,71,72. With varying protocols to generate iPSC-derived microglia, there lacks a consensus as to which of these protocols produce the most authentic in vivo microglia. For iPSC-derived microglia to be a viable option for treating neurodegenerative diseases, transplanted microglia should resemble their healthy in vivo counterparts regarding their morphology, transcriptomes, and function.
Characteristics of Microglia for Transplantation
Microglia for transplantation should be easy to access, manipulate, and have representative ontogeny and characteristics of healthy microglia found in vivo. Exogenic microglia must recapitulate a normal, healthy phenotype to supplement dysfunctional microglia or replace a lack of microglia. Characteristics of healthy microglia that should be imitated for successful exogenic microglia transplantation include normal genomic and transcriptomic signatures; morphology; phagocytic ability; participation in synaptic pruning, neurogenesis, and angiogenesis; expression of microglia-derived cytokines and chemokines; and overall transplantability (Fig. 2).

Characteristics necessary for microglial transplantation. The following are characteristics of exogenic microglia that must be considered for their success as a therapeutic. Many interacting characteristics are necessary for successful engraftment and migration of exogenic microglia in the brain. Exogenic microglia must mirror in vivo morphology from an ameboid to ramified shape. These cells must have phagocytic functionality. Exogenic microglia should express comparable genomic and transcriptomic signatures compared with their in vivo counterparts. Exogenic microglia should possess the ability to prune synapses and neuronal connections necessary for neurogenesis, as well as aid in angiogenesis. Exogenic microglia should have the ability to produce and secrete similar microglia-derived cytokines and chemokines. The ability to successfully transplant or inject exogenic microglia into the CNS must be considered, as these transplanted cells may not survive after transplantation.
Morphology
Microglia begin with an ameboid morphology during fetal development. The ameboid shape allows for motility and phagocytic functions that are necessary for apoptosis and pruning of surrounding neuronal cells 73 . As microglia continue to develop, they retain their ameboid morphology as they migrate through white and gray matter tracts. Once in the gray matter tracts, microglia undergo morphological changes to a typical highly ramified morphology seen in adult microglia 74 . The branched morphology lends microglia to maximize their ability to surveil and protect the brain 73 .
Several studies have characterized morphology and surface markers of in vitro cultures of microglia derived from iPSCs. MLCs derived from human pluripotent stem cells displayed yolk sac embryoid bodies that resembled hemogenic epithelium or the nascent blood islands observed early in microglia development. The iPSCs developed an MLC precursor phenotype of round, vacuolated cells with a compact nucleus, filopodia, and membrane ruffles. The MLC precursors expressed transcription factors PU.1, ionized calcium–binding adaptor molecule 1 (IBA1), and surface markers like cluster of differentiation (CD)11b and CD45. After further differentiation into MLCs, the purinergic receptor P2RY12, transmembrane protein TMEM119, IBA1, and CD45 were expressed 63 . Another published protocol produced MLC precursors that expressed hematopoietic progenitor surface markers CD45, CD34, and CD43. The fully differentiated MLCs expressed CD11b, CD39, CX3CR1, IBA1, CD45, and major histocompatibility complex (MHC) II cell surface receptor (HLA-DR). The MLCs underwent similar morphological changes to normal microglia, from a more ameboid to the ramified morphology seen in healthy adult microglia 75 . The presence of similar immunohistological markers provides additional evidence that these MLCs can be characterized like healthy adult microglia. A caveat to in vitro differentiation of MLCs is that culture conditions vary among these studies, allowing for differentially expressed proteins and characterizations of these exogenic microglia. In addition, the transition from ameboid to ramified in culture is similar but not identical to the normal sequence in vivo, which may impact the phenotype of MLCs71,75.
Blastocyst complementation is a possible source of exogenic microglia and may permit better recapitulation of the ameboid to branched transition due to the in vivo developmental context76,77. The potential of the use of this technique is explored in more detail in the “Generating Exogenic Microglia” section.
Genetic and transcriptomic signatures
Exogenic microglia often are maintained in tissue culture prior to transplantation. Primary microglia or microglia-derived iPSCs maintained in tissue culture then isolated and transferred to tissue culture have been shown to lack microglia-specific genes like AP-1, MEF2, MAF, RUNX, and IRF family members, as well as SMAD3 and SPI. Within 6 h these cells undergo changes in proliferation and phagocytic capacity compared with their typical healthy in vivo counterparts61,78,79. When mouse microglia cell lines and primary microglia were cultured with transforming growth factor (TGF)-β1, there was a shift in their microglial molecular signature that made them more similar to freshly isolated adult mouse microglia phenotype. The TGF-β1 cultured mouse microglia showed a more homeostatic microglial signature 80 . A deviation from the normal microglia transcriptome is problematic, as the cultured cells may no longer behave like healthy microglia and thus may be less effective upon transplantation. An additional study demonstrated that healthy, normal exogenic primary mouse microglia transplanted into a mouse CNS can return to a homeostatic state observed in healthy microglia 61 . Another study found that MLCs derived from human ESCs and iPSCs displayed a similar transcriptome to isolated human fetal microglia cultured in a neural precursor medium 63 . These studies suggest that specific culture conditions may allow MLCs to express a similar transcriptome to that of primary microglia cultured ex vivo.
The issue of mimicking a healthy and normal transcriptome is further complicated by recent studies demonstrating how regional diversity and aging influence transcripts. Regionally specific microglia in the brain have unique genetic and transcriptomic signatures in addition to that of the core transcriptome 63 . Aging mice display patterns of upregulation and downregulation of certain microglial genes as they aged from 2 to 12 months 81 . In addition, key microglial genes have been identified like CX3CR1, which plays a role in synaptic pruning, synaptic maturation, and synaptic plasticity 82 . Young CX3CR1-deficient mice display similar transcriptomic signatures of aging CX3CR1-sufficient mice, providing further evidence that transcriptomic alterations are relevant to microglial development 83 .
There is growing evidence for transcriptional differences in AD mouse models compared with human AD patients. Transcriptional profiling of microglia isolated from postmortem AD brains revealed human-associated microglia with minimal overlap with previously characterized mouse disease–associated microglia gene modules84–86. Historically, microglial activity in mouse models has been characterized by short-term, highly active innate immune responses, rather than a gradual loss of activity and senescence seen in aged human microglia 87 , which likely contributes to the species-specific differences observed between human and mouse microglia. These studies suggest that genetic and transcriptomic profiles of microglia are dynamic and can be influenced by culture methods, anatomical location, development, and disease. Exogenic microglia have the potential to recapitulate the genetic and transcriptomic signatures appropriate for a specific brain region and may be age-matched to encourage successful engraftment and migration within the transplanted brain.
Phagocytosis
While microglia are in their healthy resting state, they partake in surveillance and display homeostatic characteristics such as expressing minimal cell surface markers, releasing cytokines and chemokines, and are not actively involved in phagocytosis17,88. Microglia in an activated state are phagocytizing debris, dead cells, pathogens, and synapses 89 . In theory, exogenic microglia transplants would recapitulate the properties of both the resting state and active phagocytic state of microglia under appropriate conditions.
It is well established that primary microglia can retain their phagocytic properties in vitro with proper culture conditions. Cultures with cellular debris or pro-inflammatory cytokines such as interferon (IFN)-γ and TNF-a can induce and enhance phagocytic activity90,91. Evaluating the phagocytic capacity of MLCs is important to establish their similarity to normal, healthy microglia. Some in vitro human ESC- and iPSC-derived MLCs have been shown to phagocytose latex beads 63 , while bone marrow–derived microglia-like cells (BMD-MLCs) have demonstrated engulfment of iron particles and Aβ92,93. Human iPSC (hiPSC)-derived MLCs also phagocytized synaptosomes and participated in synaptic pruning 71 . While it is unclear whether the ability of exogenic microglia to remove debris, dead cells, invading pathogens, and synapses is equivalent to normal, healthy microglia, retaining some phagocytic function is encouraging for the desired outcome of exogenic microglia resembling normal microglia in vivo.
Pruning and neurogenesis
Like microglia’s ability to remove debris via phagocytosis, they also engulf both presynaptic and postsynaptic components and play a role in neuronal pruning89,94. Most pruning occurs in development, facilitating functional and mature neuronal circuits; although, pathological phagocytosis of synapses has also been observed34,94,95. Mice deficient in CX3CR1, a mediator of pruning, demonstrate reduced amounts of microglia and impaired synaptic and neural network connectivity96,97. Microglia help determine the connections that are conserved and maintained within the brain.
In addition to eliminating connections, the physical contact between microglia and neurons and the release of various neurotrophic factors create a cellular milieu that allows the formation of circuits and connections 98 . Microglia release factors such as brain-derived neurotrophic factor and insulin-like growth factor I (IGF-I) that promote proliferation, differentiation, and development of neural stem cells (NSCs) to intermediate progenitor cells99–101. Phagocytosis of synapses and apoptotic cells creates space and a more supportive environment for neurogenesis. Due to the significant influence of microglia on the regulation of neural networks and synapses, it is critical for exogenic microglia to maintain these functions when transplanted.
Angiogenesis
Another role of microglia is the facilitation of angiogenesis within the CNS. During embryogenesis, microglia migrate into the CNS around embryonic day 9.5 (E9.5) before vessels are present within the embryo102,103. Vessels eventually sprout, extend, branch, and anastomose with one another. As the CNS develops, the newly branched vessels become affiliated with microglia and other glial cells. There is evidence that microglia play a role in the connection of vascular sprouts during angiogenesis. In mouse embryonic hindbrains deficient in macrophages and microglia, reductions in the number of microglia were correlated with reduction in the number of branch points. Existing branch points or connections were often in contact with the neighboring vascular sprout cells when microglia were present 102 . The presence of microglia significantly increased vascular branches emanating from an aortic ring. This effect was greater when microglia made direct contact with the ring rather than in the presence of only microglia-conditioned media 104 . These findings suggest that direct cellular contact with microglia can enhance angiogenesis.
In addition to encouraging vascular development in embryogenesis, microglia can promote angiogenesis in neurologic injuries and diseases. An ex vivo assay of angiogenic ability showed that co-culturing rat primary retinal microvascular endothelial cells and microglia resulted in an increase in the release of angiogenesis-related factors in retinal microvascular endothelial cells and induced tube formation 105 . Angiogenic tube formation of endothelial cells in vitro was found in the presence of IL-4–stimulated BV2 mouse microglia cell line, and microglia promoted angiogenesis in a mouse model of stroke via secretion of angiogenic factors within exosomes 106 . These reports are representative of how microglia can promote angiogenesis in both normal and injured or diseased brains. Pro-angiogenic function may be an important characteristic to consider when assessing exogenic microglia for transplantation, especially for treating brain disorders involving vasculature.
Microglia-derived cytokines and chemokines
As aforementioned, microglia produce both pro-inflammatory and anti-inflammatory cytokines and chemokines. Pro-inflammatory factors can encourage neurotoxic states, while anti-inflammatory factors promote healing 107 . For example, when microglia are induced by TLR and IFN-γ signaling pathways they produce pro-inflammatory factors, including TNF-a, IL-6, IL-1β, IL-12, chemokine ligand 2, nitric oxide (NO), matrix metallopeptidase (MMP)-12, MHC class II, Fc receptors, and integrins107–110, while the anti-inflammatory factors released from microglia include: IL-10, TGF-β, IGF-I, fibroblast growth factor, colony-stimulating factor (CSF)-1, neurotrophic factors, and progranulin107,111. The variety of secreted cytokines and chemokines from microglia has a broad influence on the CNS environment through neuroinflammation and neuroprotection.
The production and release of secreted factors must be maintained in exogenic microglia, and it important that these factors are released at the appropriate time, place, and quantity. Specifically, pro-inflammatory cytokines and chemokines must be maintained at normal, healthy levels in exogenic microglia transplants to permit proper function yet avoid contributing to chronic inflammatory diseases of the CNS. Similarly, the secretion of anti-inflammatory cytokines and chemokines by exogenic microglia should emulate that of normal, healthy microglia to allow alleviation of inflammatory CNS disease pathology and symptoms, while not compromising the recipients’ ability to fight acute infection. An additional consideration is that cytokines and chemokines released by exogenic microglia may have off-target effects via paracrine signaling. The impact of the exogenic microglia–derived cytokines and chemokines would need to be fully addressed regarding the relevant disease pathology and symptoms.
Transplantability of microglia
Function notwithstanding, transplanted microglia must demonstrate consistently the ability to engraft and survive in the recipient. Numerous studies have shown that transplanted microglia engraft and spread into surrounding brain regions61,64,71. Notably, extensive engraftment of transplanted microglia was observed in mice with homozygous knockout (KO) of colony-stimulating factor 1 receptor (CSF1R), which regulates macrophage lineage cells (eg, microglia) proliferation, differentiation, and activation61,112. Selected studies have demonstrated the transplantability of exogenic microglia for a variety of diseases and injuries including AD and SCI. Transplanted microglia recapitulated important functions of normal microglia including phagocytosis, migration, and anti-inflammatory signaling64,71, suggesting that transplantation is feasible, but the degree of transplantability is still actively being assessed. One major challenge arises from the responsive nature of microglia, since environmental factors drive their function. For example, if exogenic microglia are transplanted into a neurodegenerative brain, exogenic microglia potentially could adopt the characteristics of the already present diseased microglia due to the inflammatory milieu. An additional challenge for microglial transplantation, as with other cellular and organ transplants, is avoiding immune rejection of the grafted tissue by the recipient. While the CNS was once thought to be “immune-privileged,” it is now established that transplantation into the brain or spinal cord evokes a host immune response. Both the adaptive and innate immune systems appear to contribute to graft rejection, whereas MHC matching and the use of undifferentiated iPSC may reduce, but do not eliminate, immunogenicity of allogeneic grafts113–116. Many preclinical transplantation experiments, including those reviewed here, utilized immune-deficient recipient animals and donor microglia syngeneic to the recipient.
For microglia transplantation to be applied for the treatment of human disease, the use of autologous or highly donor-matched cells may reduce the need for immune suppression. Xenotransplantation, or the transplantation of tissues from different species, has been explored as an alternative approach, and has become a topic of increased public awareness after the transplant of pig heart tissue into a human patient at the University of Maryland 117 . For this strategy to be applied to microglia, it is likely that genetic engineering to “humanize” the tissue, perhaps in addition to immune suppression, would be necessary. In addition, species-specific differences in microglial genetics, morphology, and function between humans and other species would need to be considered.
Other strategies to avoid immune rejection of transplanted tissues which may be applied to microglia transplantation include encapsulation and the creation of “universal” pluripotent cell lines. In an encapsulation approach, the donor tissue is surrounded with a biocompatible, selectively permeable barrier 118 . Encapsulation may only be appropriate for certain microglial transplant approaches, that is, if the proposed therapeutic mechanism is the secretion of neurotrophic factors. As the barrier prevents cell–cell contact, it would preclude phagocytic activity of the transplanted cells. So-called “universal” pluripotent cells are typically genetically modified to KO HLA and knock-in inhibitors of natural killer cells, which would typically recognize lack of HLA molecules 119 . The use of MLCs derived from “universal” pluripotent cells may be a potential approach to improve graft integration. A microglia-specific strategy is a depletion then repopulation approach. Studies have shown that after depletion of brain microglia, circulation-derived cells which resemble microglia repopulate the brain; however, further research is necessary to characterize the effects of transient depletion, and whether circulation-derived cells adequately recapitulate microglial function in the long-term120,121.
Species-specific differences in microglia
The percentage of microglia found in the human and mouse brain varies with microglia making up 0.5–16.6% of the total cell population in the human brain and 5–12% in the mouse brain122,123. Although the percentage of microglia is similar, it is unclear how this difference impacts the capacity of the recipient animal to support transplanted microglia.
Both mice and human microglial cells found in the CNS can be distinguished from peripheral monocytes and other immune cells based on their transcriptional signature 80 . Mice and human microglia also have regional variations suggesting species-specific microglial diversity124,125. It is unknown if this microglial diversity is translatable from one species to the other. When comparing mouse and human microglia’s sensome, a set of highly expressed genes in sensing pathological conditions, there were 57 genes that overlapped and considered core microglia sensome genes 126 . The similar genes will need to be verified through other means, but the study provides insight on genes that are found within both species. Significant differences have been reported in microglial genes, with only about half of the mouse microglial signature overlapping within humans 79 , while another study reported differences in human and various mouse microglial gene data sets ranging between 9% and 20% 127 . Variation exists between human and microglial transcriptomes, and thus must be considered for characterization studies of in vivo human microglia developing in a mouse host. The in vivo microenvironment may alter the transplanted human microglia gene expression profile. Future transplantation studies should characterize the transplanted microglia in comparison with the intended recipient species to see if they are similar.
Microglial Transplantation Studies
Recent studies of cell-based therapies aimed at treating CNS disorders suggest that transplantation of MLCs can ameliorate pathology and restore neurological function 120 . Transplantation of microglia derived from human ESCs into mouse brains has demonstrated successful engraftment of microglia that are transcriptionally comparable with primary microglia found in the mouse brain 128 . In addition, transplanted immortalized mouse and primary rat microglia have led to neuroprotection by promoting angiogenesis and regeneration, suppressing inflammatory responses, and reversing behavioral deficits106,129. These breakthroughs in transplantation research suggest that the grafting of young and healthy microglia would be a novel approach for treating neurological disorders with microglial dysfunction (Table 1).
Animal Studies of Microglial Transplantation.

BMD-MLC: bone marrow–derived microglia-like cell; CSF: colony-stimulating factor; eGFP: enhanced green fluorescent protein; GDNF: glial cell–derived neurotrophic factor; GM: granulocyte-macrophage; HPC: hematopoietic progenitor cell; HSC: hematopoietic stem cell; IL: interleukin; iMLC: induced microglia-like cell; MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; SCI: spinal cord injury; TBI: traumatic brain injury; WT: wild-type.
Microglial Transplantation for AD
While the most common and significant risk factor for AD is age, the innate immune system has been implicated in the onset and progression. Genetic studies in the early 2010s identified many AD risk genes associated with microglial activation142–144. Variants in the gene triggering receptor expressed on myeloid cells 2 (TREM2) are associated with microglial activity and are correlated with AD disease145,146. Microglia have been shown to cluster around senile plaques and phagocytose amyloid147–149. In addition, analysis of TREM2 expression in AD mouse models suggests that a loss of microglial function increases the risk of AD150–155. On the other hand, microglia have been associated with activities contributing to the progression of AD, such as phagocytosis of synapses, mediating the spread of tau pathology, and the secretion of pro-inflammatory factors156–159. In line with these results, other studies have observed that deficiency in TREM2 is protective against neurodegeneration160,161.
Recent work suggests interactions with other immune system components may be responsible for the opposing roles that microglia play in the progression of AD associated with Aβ plaque deposition, astrogliosis, and microgliosis in relation to C3 complement protein11,38,60,162–164. In addition, microglia appear to lose phagocytic capability over time, and this effect may be compounded with age, chronic activation, and the progression of AD pathology. Microglial activation may initially be protective in AD, but an accumulation of risk factors, such as genetics, stress, metabolism, age, and amyloid and tau pathology, disrupts microglial homeostasis, causing microglia to become senescent and harmful. Recent in-depth reviews describe the role of microglia in AD86,143,165,166.
Conflicting evidence on whether microglia can protect or exacerbate the progression of AD leads to the necessary examination of the potential safety and efficacy of microglial transplants as a potential therapy for AD. Microglia can reduce plaque burden via phagocytosis and sequestration of amyloid167–169. There is evidence that stimulation by external signals, such as cytokines and opsonizing antibodies, enhances phagocytosis of Aβ170–172. In addition, microglia modulate host immune functions and support host neurons in early development through the secretion of neurotrophic factors, facilitating the survival of neurons in postnatal development 173 . Transplantation of exogenic microglia may provide benefits for AD by slowing or preventing Aβ pathology as well as supporting host neurons.
On the other hand, microglia may play a role in driving AD, particularly microglia with atypical phenotypes. Aβ deposition and plaques can induce microglia into a chronic or hyper-activated state in which they produce pro-inflammatory cytokines and toxic compounds like NO174,175. While microglia can phagocytose Aβ this “activated” phagocytic state may be induced by chronic inflammation34,157,176. Both aging-associated and AD-associated microglia are at risk for deterioration and display reduced motility and phagocytosis compared with their healthy counterparts162,177. This evidence suggests that exogenic microglia may acquire detrimental functions upon transplantation into an aged, inflammatory environment and aggravate rather than treat AD pathology.
Several preclinical studies have investigated microglial transplantation in AD rodent models. Exogenous microglia have been shown to migrate throughout the brain, while transplanted microglia co-localize with Aβ deposits in conjunction with endogenous immune cells. In this same study, TNF-a levels were elevated in rats that received transplantation, which the authors interpreted as an increase in microglial activity 130 . It was uncertain whether Aβ clearance in rats that received microglial transplantation was solely due to direct activity of exogenous microglia, or whether the transplanted microglia recruited endogenous immune cells, neither was it clear if whether transplanted microglia survived and retained beneficial function long-term. Another in vitro microglia study suggested that soluble factors secreted by young microglia can restore old microglia to a phenotype which counteracts amyloid pathology 178 . Co-culturing brain slices or conditioned media from wild-type (WT) neonatal mice and aged APP/PS1 mice resulted in approximately 60% reduction of overall amyloid load in the AD slices. These results indicated that both young and old microglia were responsible for amyloid clearance, particularly old microglia. This is promising for the prospect of microglial transplant as it suggests that exogenic microglia maintain their identity and can ameliorate the diseased milieu of the host, rather than adopting the inflammatory environment.
Despite promising results using neonatal microglia transplantation in a mouse model of AD, there are many ethical and technical challenges with the procurement of primary human microglia. Some recent studies have focused on characterizing the transplantation of BMD-MLCs for AD131,132. Mouse bone marrow cells were harvested and differentiated into cells with a microglia-like phenotype by culture with CSF-1/macrophage-CSF. These BMD-MLCs steadily expressed microglia-associated biomarkers such as CD11B, CD68, IBA1, and TREM2 and demonstrated capacity to phagocytose Aβ directly and induce primary microglia to phagocytose Aβ in culture92,93. BMD-MLCs showed a mild inflammatory cytokine profile compared with primary microglia and macrophages, with reduced secretion of IL-β1 and IL-6 but comparable secretion of TNF-a in pro-inflammatory conditions 131 . The secretion of TGF-β1 by BMD-MLCs was observed to stimulate phagocytic activity of host microglia 132 . BMD-MLCs transplanted into the hippocampus of APdE9 mice engrafted, migrated, and co-localized with Aβ deposits throughout the brain at 7 and 14 days post-transplantation. The total number of Aβ plaques decreased at 14 days post-transplantation and correlated with improvement in performance on the Novel Object Recognition test 131 . These findings suggest that BMD-MLC transplantation resulted in a short-term reduction in amyloid burden and improved cognition in a mouse model of AD.
Other studies have investigated the potential of iPSC-derived microglia for transplant in AD models. The generation of induced microglia-like cells (iMLCs) from hiPSC was first differentiated into hematopoietic progenitors with the aim of recapitulating normal microglial development. Induced hematopoietic progenitors were then further differentiated into iMLC. iMLC expressed microglia-associated proteins including PU.1, TREM2, CX3CR1, and TGFβR1, and resembled human microglia morphologically. Transcriptome analysis revealed iMLC clustered closer to human adult and fetal microglia than to either hiPSC or induced hematopoietic progenitors. iMLC displayed expected microglial functions including cytokine release and phagocytosis 71 . When iMLCs were transplanted into Rag2−/−/Ilrγ−/−:5xFAD mice, they migrated toward and engulfed Aβ plaques 71 . These results suggest that transplant of iMLC and other iPSC-derived microglia cells may be beneficial for reducing plaque burden in AD.
These studies provide hope for the effectiveness of microglial transplant to treat AD symptomatology including amyloid pathology and cognitive deficits. Further study of microglial transplantation in AD models is needed given how microglial activity in AD patients is context dependent. Particular attention should be paid to microglial phenotype and disease progression to evaluate whether microglia transplantation offers significant benefit. The source and characteristics of exogenic microglia must be considered to address concerns such as scalability and the need for high-quality products. Whether coadministration of microglial cell transplant in addition to other treatments may improve therapeutic benefits should also be considered.
Microglial Transplantation for PD
Reactive microglia in the substantia nigra (SN) have been implicated in PD since 1988, and recent genomics and transcriptomics methods have allowed significant advancements in understanding the relationship between neuroinflammation and PD179–182. Accumulation of misfolded α-synuclein is thought to contribute to PD pathogenesis through its assembly into Lewy bodies and Lewy neurites, spreading throughout the brain via cell-to-cell transmission183–187, while priming microglia with a pro-inflammatory stimulus also enhanced the accumulation of α-synuclein 188 . These results suggested that microglial activation is correlated with the progression of α-synuclein pathology in PD.
Current PD treatments are not disease-altering and relieve motor signs such as resting tremors, rigidity, or akinesia with limited efficacy. Historically, investigation of transplantation therapy for PD has focused on intracerebral transplantation of dopaminergic neurons; however, more recent studies have shifted their attention to stem cell transplantation with ESCs or NSCs116,189–191. Other cell types are being explored due to inconsistent results, unsuccessful clinical trials, and ethical and methodological concerns regarding the prospects of fetal neuron transplantation. A more novel approach is to use MLCs and macrophage-like cells as a therapy for PD, as microglia play a role in neuroinflammatory mechanisms found in PD. Microglia mediate Lewy body pathology by inducing inflammation in PD 116 , while other mechanisms include the microglia-associated and PD risk gene TREM2, and an inflammatory phenotype that worsens PD’s neuroinflammation192,193. The role of microglia in PD provides an avenue for the transplantation of exogenic microglia as a novel therapy. By providing healthy, exogenic microglia to replace the dysfunctional microglia in PD, the degenerative neuropathology and clinical signs may be mitigated.
A recently developed strategy used hematopoietic stem cell (HSC) transplantation–based macrophage-mediated glial cell–derived neurotropic factor (GDNF) delivery to home HSC-derived macrophages to the sites of neurodegeneration133,134. Mice treated with neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) developed a PD-like phenotype including dopaminergic neurodegeneration. Delivery of the GDNF-expressing HSC-derived macrophages to MPTP mice rescued tyrosine hydroxylase-positive neurons of the SN and terminals in the striatum and stimulated axon regeneration133,194. Macrophage-mediated delivery recovered general ambulatory decline found in PD-like animals 133 . GDNF-expressing HSC-derived macrophages transplanted into MitoPark mice exhibiting PD-like signs have shown transgene-expressing macrophages infiltrating degenerating CNS regions and increased GDNF levels in the midbrain of the PD-like mice 134 . These studies suggest that GDNF-expressing HSC-derived macrophages can be transplanted successfully to promote a neuroprotective environment for PD individuals. However, one caveat to these studies is that macrophages are delivered prior to inducing the PD-like state, and further research is needed to investigate this approach as a therapeutic rather than preventative.
Microglial Transplantation for Stroke
There are not many widely used therapies for secondary injury induced by stroke. The current cell transplantation therapies show promising results in negating these secondary effects in stroke animal models. These therapies include mesenchymal stem cells (MSCs), NSCs from various sources and derivations and subjected to various pretreatment, and extracellular vesicles (EVs) derived from MSCs and microglia135,195,196. Each of these stem cells has demonstrated a therapeutic relief for stroke models, and recent research has found that microglia derivatives like EVs can alleviate stroke symptoms135,197.
EV treatments for stroke are often derived from MSCs, but more recent studies have begun to explore EVs derived from microglia. Transplantation of EVs derived from microglia has shown promise in the stroke recovery process. Regeneratively stimulated EVs, like IL-4, were derived from WT mice that were washed with ATP (to promote EV development) and treated with pro-regenerative conditions. These vesicles were shown to promote beneficial functions of microglia and macrophages and prevent immune cell deterioration in poststroke oligodendrocyte precursor cells to enhance remyelination of neurons in mouse models135,198. While EVs provide one method for transplantation, more conventional cellular transplantations are also being evaluated.
Transplantation of microglia has been shown to reverse some of the damage caused by stroke. Transplanted microglia from postnatal mouse primary cell cultures that were preconditioned by oxygen–glucose deprivation (OGD) improved neurological deficits scores in comparison with a postnatal mouse primary cultured, OGD-preconditioned astrocyte group and a control group. Improvement in functional recovery was compared with a normoxia-preconditioned microglia transplantation group. The rats that were transplanted with OGD microglia also showed an increase in levels of growth factors MMP-9 and vascular endothelial growth factor 136 , which is beneficial considering one characteristic pathology of any CNS injury is vascular disruption. These findings suggest that OGD microglia induced changes in the activation of resident microglia and promoted angiogenesis and axonal outgrowth, significantly improving some of the deleterious outcomes of stroke in rodent models. An additional study using a human immortalized microglial cell line HM06, created by isolating microglia from human fetal telencephalon tissue, found that transient middle cerebral artery occlusion (MCAO) rats transplanted with HM06 displayed improvement in neurological recovery, decrease in infarct volume, and fewer apoptotic cells in the ischemic core and penumbra 137 . Taken together, these studies suggest that transplanted microglia appear to have substantial effects on the reduction of damage and improvements in functional and cognitive recovery in rodent models of ischemic stroke.
Another study performed on rats compared the effects of transplantation of bone marrow mononuclear cells and microglia after permanent MCAO and found no neuroprotective results after microglial transplantation. There was no evidence of a reduction in brain water content at 3 days post injury (DPI), or an effect on infarct volume and neurological function up to 14 DPI 138 . Several factors may impact the success of microglial transplantation therapy such as the source of microglia, the type of injury, and timing of transplantation. These findings may also be due to different stroke models and methodologies, yielding significant differences in results compared with more recent studies that have demonstrated the restorative properties of microglial transplantation in ischemic stroke models. With only a few studies assessing the outcomes of microglial transplantation in stroke models, this avenue of research should continue to be explored.
Microglial Transplantation for SCI
As the neuroprotective role of microglia has become more established within the context of SCI, researchers have turned to examine the potential therapeutic benefit of microglial transplantation to restore nervous system function. Three key studies64,139,140 outlined the subtype-specific and time-dependent restorative effects of microglial transplantation after SCI with respect to motor function and tissue preservation.
Kou et al. directly transplanted microglia into the rat spinal cord in the subacute stage of SCI and analyzed motor function and tissue integrity as markers for CNS restoration. The rats received either a microglial transplant or a control saline injection 1 week post-injury (WPI). Primary microglial cells were dissected and isolated from postnatal rats and allowed to proliferate in vitro prior to transplantation. The researchers reported significantly improved motor function in microglia-transplanted rats as measured by various behavioral assessments. Microglia-transplanted rats also showed a decrease in latency of motor-evoked behavior and a reduction in tissue damage and lesion size in response to microglia transplantation as compared with saline control 139 .
Akhmetzyanova et al. provided results suggesting that M2-specific microglial transplantation immediately after the acute stage of SCI promoted greater tissue preservation but did not significantly improve motor functional deficits. Like as described in Kou et al., rats received the same type of injury; however, microglial transplantation occurred immediately after injury. This did not translate into an improvement of motor function, but M2 microglia transplanted in the acute stage of SCI appear to promote preservation of tissue integrity but do not improve motor deficits 140 .
In a mouse model for SCI, Kobashi et al. transplanted microglia into the mouse spinal cord immediately after injury to assess the extent of functional recovery. The microglia were activated separately into pro-inflammatory M1 and anti-inflammatory M2 microglial phenotypes by introduction to granulocyte-macrophage CSF or IL-4, respectively. M2 microglia–transplanted mice displayed a significant restoration of motor function 4 WPI as compared with M1-transplanted mice and controls as measured by Basso Mouse Scale and hindlimb reflex scoring. In addition, in M2 microglia–transplanted mice alone, the authors described a recovery of retrograde axonal transport 64 , ultimately providing evidence for potential restorative effects of subtype-specific microglial transplantation.
Microglial Transplantation for TBI
Acute manifestations of TBI can be resolved if microglia do not remain activated; however, there is evidence of chronic inflammation caused by sustained microglial activation that leads to cognitive decline4,159,199,200. The attenuation of neurological dysfunction can be achieved by regulating microglial function 139 . Various cell types and cell-based therapies have been explored as potential treatments for TBI. These treatments are intended to support neurological restoration, neuroprotection, and functional recovery in TBI patients. To date, the cell types tested have been primarily stem cells, including umbilical cord blood stem cells, MSCs, hiPSC-derived neural cells, and NSCs201–204. Each cell type has shown varying levels of success for treatment of TBI.
Microglia are implicated in TBI pathology and phagocytosis of transplanted cells for experimental TBI treatments; therefore, altering the microglia population of TBI patients may provide an alternative approach to developing TBI therapies. When microglia are removed from a TBI brain, there is little effect on the outcome of the injury 121 . Recent studies have focused on understanding and promoting microglia with an anti-inflammatory phenotype rather than microglia with a pro-inflammatory phenotype through pharmacological compounds and ligands to alleviate functional deficits in TBI mice194,205. Promoting the neuroprotective phenotype in a TBI mouse model stimulates adult neurogenesis and aids in recovery of cognitive function by supporting the survival of newborn neurons121,141. Encouraging microglia turnover through genetic or pharmacologic methods has shown that new mouse host-derived microglia can alleviate some TBI symptoms and pathology like improving spatial learning and memory and promoting functional neurogenesis 121 . This evidence suggests that microglia phenotypes may be relevant for treating TBI, while also demonstrating the importance of the presence of microglia in treating TBI.
Transplantation of exogenic microglia is an alternative approach for TBI therapy. By introducing regional neuroprotective microglia to an affected TBI area, exogenic microglia may provide additional support, such as neurotrophic factors or homeostatic cytokines, to primary microglia in the brain. In addition, if these exogenic microglia are patient specific, they will be less likely to elicit an immune response, resulting in less phagocytic activity by primary microglia.
These studies provide evidence that microglia transplantation can restore function in a variety of brain and spinal cord disorders. The clinical translation of this approach, however, will require sources of human microglia cells for transplantation.
Generating Exogenic Microglia
Sources of human microglia for transplantation include those derived from human fetal brain tissue, and the differentiation of hiPSCs. Ethical and logistical issues of harvesting human fetal brain tissue limit this as a source for generating human microglia. The differentiation of hiPSCs into microglia has yet to produce authentic cells that exhibit the regional diversity of microglia cells typically found in the brain.
Authentic cells can be produced via exogenesis by generating animal chimeras via the process of blastocyst complementation. This method involves the injection of donor stem cells (eg, iPSCs) into a genetically modified host blastocyst stage embryo. A target gene of interest is ablated in the host embryo to create a developmental niche. Donor stem cells are injected into the empty developmental niche, resulting in the generation of the missing organ or cell type of interest76,77. An advantage of this technique is that the donor cells and their progeny are then subject to similar spatial and temporal developmental cues as those experienced by progenitor cells in a normal, healthy embryo (eg, morphogen gradients, cell–cell contacts). Numerous blastocyst complementation studies have demonstrated the ability to generate liver, forebrain, kidney, pancreas, lung, thyroid, and eye77,206–210; however, no blastocyst complementation studies have focused on developing tools to create exogenic microglia in the brain.
Recent evidence has shown that blastocyst complementation can produce inner ear sensory neurons to correct inner ear malformations, while neural blastocyst complementation can target neuronal formation in the cerebral cortex and hippocampus211,212. While neural blastocyst complementation studies generated neurons, these studies also observed non-dorsal telencephalic progenitors such as interneurons, microglia, and blood vessel endothelial cells, demonstrating chimerism211,213. Although in this case, the chimerism of microglia was an off-target effect, demonstrating that donor cells injected during blastocyst complementation can contribute to microglia development. Previous studies have generated exogenic immune cells through blastocyst complementation with WT stem cells in murine embryos genetically engineered to KO IL2Rg, RAG2, and C-KIT genes214,215. These studies suggest that blastocyst complementation can target specifically cells in the brain as well as immune cells, supporting the fact that microglia are a reasonable target for exogenic generation via blastocyst complementation.
A critical step in the blastocyst complementation approach is understanding the development of normal microglia and, importantly, the key genes and transcription factors that are involved in the specification and development of microglia. These genes that are activated during the developmental process serve as candidate targets for KO to create niches for injected stem cells to develop into exogenic microglia.
Microglia are derived from the yolk sac around mouse E7.5 during the first wave of hematopoiesis59,67,216–223, and they begin to migrate shortly after they are generated through the vasculature system59,223. These microglial precursors enter the brain around mouse E9.5–E10.5 and rat E11–E12 and spread throughout the cortex59,220,223–228. A second wave of hematopoiesis generates Hox8b+ cells that expand in the fetal liver and aorta-gonad-mesonephros before migrating to the CNS at mouse E12.5229–232. Microglial cells are maintained separately from bone marrow mononuclear cells through their ability for self-renewal233–235. The density in microglia stabilizes after postnatal week 6223,236, postnatally transitioning from an ameboid to branched morphology237,238. As microglial cells start their journey into the yolk sac and migrate to the brain, they undergo a variety of developmental changes that are driven by expression of genes, transcription factors, morphogens, and receptors (Fig. 3).
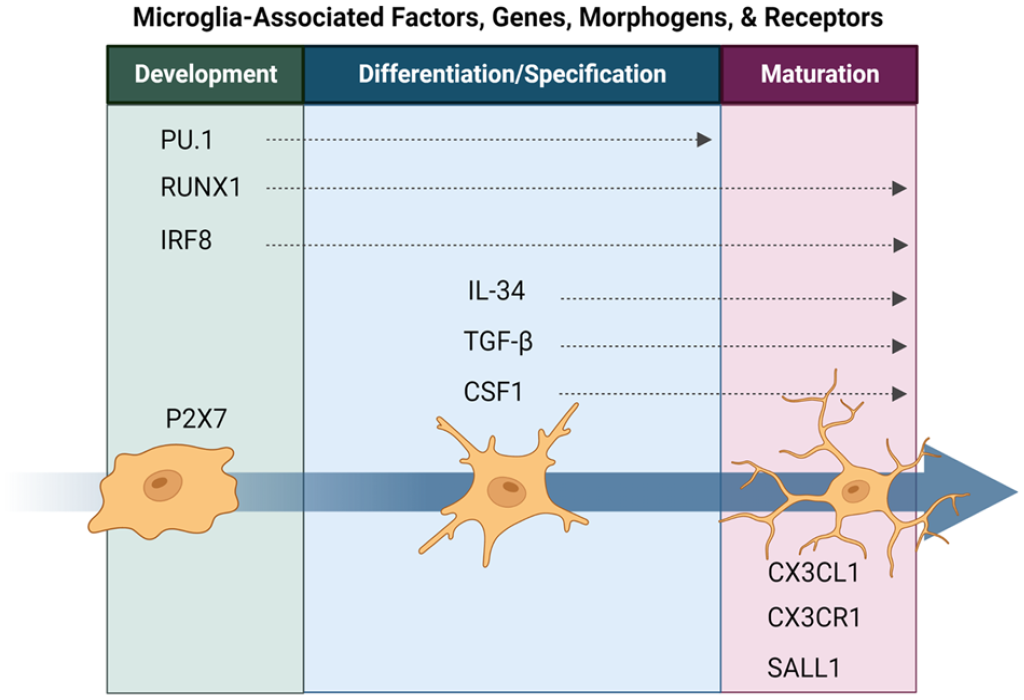
Various factors, genes, morphogens, and receptors are associated with microglial development, differentiation, and maturation. Microglia-associated factors, genes, morphogens, and receptors. Many characteristics are likely interacting with one another to promote successful engraftment and migration of exogenic microglia throughout the brain. The exogenic microglia characteristics include but are not limited to morphology, phagocytosis, genomic and transcriptomic signatures, pruning and neurogenesis, angiogenesis, microglia-derived cytokines and chemokines, and the overall transplantability of the exogenic cell.
Genes and transcription factors that play a critical role in microglia development include RUNX1, PU.1, IRF-8, and SALL1220,221. The transcription factor RUNX1 binds to regulatory elements of PU.1 and is first expressed around mouse E6.5. RUNX1-positive cells will develop into lymphoid progenitors and HSCs at mouse E7.567,221,239,240. These cells will then migrate through the vasculature and enter the brain around mouse E9.559,67. The loss of RUNX1 leads to less microglia transitioning from ameboid to ramified morphology59,67.
The transcription factor PU.1 is constitutively expressed in resting and active microglia158,241. PU.1 has been implicated in differentiation and self-renewal microglial progenitors that influence myeloid lineage commitment220,242. Null and deficient PU.1 mice lack macrophages and microglia243,244, impeding yolk sac myeloid progenitor maturation 245 .
The transcription factor IRF-8 is involved in microglial cell homeostasis and is considered a master regulator of microglial identity 246 ; however, there were mixed findings observed in IRF-8-deficient and null mice. Some studies suggested a greater abundance of microglia and changes in morphology 247 , while others observed decreased parenchymal microglia and increased apoptosis of microglial progenitors221,248,249. In addition, the zinc transcription factor SALL1 expressed in microglia is driven by TGF-β and is important for microglial maturation and homeostatic phenotype maintenance of cultured microglia220,250–252. These transcription factors are one facet in microglial development.
Morphogens and receptors provide another facet of microglial biology that impacts developmental changes observed in the microglial cell population. As previously mentioned, TGF-β is important for microglial maturation induced by the upregulation of SALL1250,251 and microglial survival and differentiation78,80,223. The purinergic ionotropic receptor P2X purinoceptor 7 is expressed in embryonic microglia and mediates further proliferation of the microglial cells 253 . Some receptor–ligand pairs also influence microglial developmental biology. CX3CL1 and CX3CR1 are important for postnatal microglial and neuronal signaling and function96,97. The CSF1R expressed on mononuclear myeloid cells and ligand IL-34 regulate microglial functions, such as phagocytosis, chemotaxis, survival, and differentiation78,247,254,255. Mice with a CSF1R frameshift mutation lack tissue macrophages but have normal microglia256,257. CSF1R null mice lack microglia entirely 258 , suggesting that CSF1R is essential in microglial differentiation.
Each gene, transcript, morphogen, and receptor play substantial roles in microglia development, including proliferation, differentiation, maturation, and survival of the microglial population. These developmental factors provide reasonable targets for generating exogenic microglia (Fig. 3).
Translation of Exogenic Microglia to the Clinic
The senescence and dysfunction of microglia are fundamental features of various neurodegenerative disorders and contribute to the exacerbation of the disease process. The replacement of dysfunctional microglia in several animal models of neurological disorders and subsequent restoration of neurological function offer great hope for treating these degenerative disorders of the brain. AD has the greatest prevalence among all neurodegenerative diseases; therefore, microglia transplantation would have the greatest societal impact in treating these patients.
However, the first attempts for microglia replacement therapy in the clinical setting will be those disorders that result in fatal outcomes within finite time periods after disease diagnosis. Therefore, the most likely candidates for treatment with microglia replacement therapy will be patients with Huntington’s disease (HD). The genetic mutations underlying HD are well characterized and as stated previously involve the trinucleotide repeat of the huntingtin protein. Since onset of HD can be predicted by number of trinucleotide repeats exhibited in the huntingtin gene, this can serve as a biomarker for stratifying patients to determine the efficacy of microglia transplantation in delaying the onset of the disease process as a primary outcome measure.
Recently, the elimination of microglia in the R6/2 transgenic mouse model of HD using CSF1R inhibitor PLX3397 resulted in improved cognitive and motor function in these animals along with reduced neuropathology 259 ; however, the consequences of the re-expansion of endogenous mutant microglia following the cessation of PLX3397 therapy were not investigated. This issue was addressed in a murine model of cerebellar Purkinje cell degeneration where microglia depletion by PLX5622, and analog of PLX3397 120 . Following the cessation of PLX5622 treatment exogenous HSCs were administered systemically to replace endogenous mutant microglia. This approach for microglia replacement with non-mutated cells prevented the loss of endogenous Purkinje cells and restored motor function, and provides a strategy for translating this approach to the clinic.
Conclusion
Microglia play a critical role in brain injuries and neurodegenerative diseases through neuroinflammation and neuroprotection. The wide range of neuroinflammatory and neuroprotective functions allows microglia to protect the CNS from injuries and diseases when the brain is healthy. Under injured or diseased conditions microglia may take on additional roles that are detrimental to the CNS. Technologies and therapeutics that intervene with the maladaptive roles of microglia may alleviate degenerative neuropathology and symptoms seen with a given injury or disease. Transplantation of healthy exogenic microglia may provide such a therapeutic approach. Several characteristics to consider when developing exogenic microglia include morphology, phagocytosis, genomic and transcriptomic signatures, pruning, participation in neurogenesis, angiogenesis, and the overall transplantability. Authentic exogenic microglia can be generated through blastocyst complementation if targeted genes for KO in embryos are carefully selected. The generation of exogenic microglia will provide a novel, more accessible, and renewable source of microglia compared with conventional donor microglia.
Footnotes
Authors’ Contributions
Conceptualization—S.R.V., P.S., and W.C.L.; literature search and data curation—S.R.V., P.S., S.T.J., A.R., and Z.V.; writing – original draft preparation—S.R.V., P.S., S.T.J., A.R., and Z.V.; writing – review and editing—S.R.V., P.S., and W.C.L.; visualization—S.R.V. and P.S.; supervision—S.R.V. and P.S.; project administration—W.C.L.; funding acquisition—W.C.L. All authors have read and agreed to the published version of the manuscript. Susanna R. Var and Phoebe Strell contributed equally to this work.
Ethical Approval
This study was approved by our Institutional Review Board.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by the NIH grant (R01 AI173804), and by the Suzanne M. Schwarz Fund.