
Abstract
Alzheimer’s disease (AD) is a devastating neurodegenerative disease with limited therapeutic options. Cellular transplantation of healthy exogenic neurons to replace and restore neuronal cell function has previously been explored in AD animal models, yet most of these transplantation methods have utilized primary cell cultures or donor grafts. Blastocyst complementation offers a novel approach to generate a renewable exogenic source of neurons. These exogenic neurons derived from stem cells would develop with the in vivo context of the inductive cues within a host, thus recapitulating the neuron-specific characteristics and physiology. AD affects many different cell types including hippocampal neurons and limbic projection neurons, cholinergic nucleus basis and medial septal neurons, noradrenergic locus coeruleus neurons, serotonergic raphe neurons, and limbic and cortical interneurons. Blastocyst complementation can be adapted to generate these specific neuronal cells afflicted by AD pathology, by ablating important cell type and brain region–specific developmental genes. This review discusses the current state of neuronal transplantation to replace specific neural cell types affected by AD, and the developmental biology to identify candidate genes for knockout in embryos for creating niches to generate exogenic neurons via blastocyst complementation.
Keywords
Introduction
Neurological Deficits in Alzheimer’s Disease
Alzheimer’s disease (AD) is a neurodegenerative disorder with a complex genetic component. The heritability of AD has been estimated to be between 60% and 84%; however, all the established genetic factors to date explain less than half of this heritability
1
. AD is primarily associated with aging and characterized by gradual loss of cognitive function. Clinical signs and their onset can vary on an individual basis, which makes diagnosis challenging
2
. The most common neurological deficit observed is progressive memory loss. Affected individuals may also experience impairment of higher order functions, struggle with planning and problem-solving, lack awareness of time and location, and experience difficulties with everyday tasks. Changes in mood, anxiety, withdrawal, and depression may also occur
3
. These changes are distressing to those afflicted and extend the burden of disease. Developing treatments for AD has been less than fruitful in recent decades. Historically, available pharmacological agents have included cholinesterase (ChE) inhibitors—galantamine, rivastigmine, donepezil—and N-methyl-
One such solution is the replacement of degenerating neurons by transplantation. Numerous studies have evaluated the transplantation of various neuronal phenotypes in animal models of AD, and demonstrate the integration of the transplanted cells into the host brain circuitry and associated restoration of neurological function. The source of these neurons for transplantation in the initial studies was neuronal progenitor cells of the appropriate phenotype derived from the fetal brain. For clinical translation of neuronal transplantation for treating AD, sources of cells other than from the human fetal brain will be required.
Various neuronal phenotypes have been generated from primary fetal cells6,7, generated from somatic cell nuclear transfer8–10, and derived from induced pluripotent stem cells (iPSCs)6,7,11,12. All of these methods offer possible alternative sources of neuronal cells for transplantation, but they lack the expression of the complete repertoire of genetic programs needed for the generation of authentic neurons 11 . Some of these methods are also expensive and some raise ethical concerns related to the origins of the transplanted cells. While blastocyst complementation provides an alternative approach to generating neuronal cells in vivo. An in vivo context encourages the expression of similar genetic programs and developmental cues to recapitulate functional neuronal cells via blastocyst complementation (Table 1). Blastocyst complementation employs targeted gene knockouts in the developing embryo/blastocyst, which creates a developmental niche for wild-type stem cells to occupy and develop into the desired cell phenotype. The resulting animal is a chimera with donor stem cell–derived tissue, and the donor-derived tissue can be specified by careful selection of the target gene. Several interspecies chimeric barriers still exist for this method12–14. However, blastocyst complementation remains a promising and innovative method for generating a variety of tissue-specific exogenic cells with the potential to treat a variety of diseases including AD.
Advantages and Disadvantages of Exogenous Neuron Sources.

iPSC: induced pluripotent stem cell.
Generating Exogenic Cells and Organs via Blastocyst Complementation
Blastocyst Complementation
Blastocyst complementation provides an innovative approach to developing a renewable source of organs and cells for degenerative diseases. Organ and cell generation via blastocyst complementation involves the injection of donor stem cells into a genetically modified host blastocyst stage embryo. The host embryo is genetically modified to ablate a gene important for a particular organ’s development, creating a developmental niche that results in agenesis of the organ of interest 12 . The injected donor stem cells, which are not genetically modified, and thus possess the necessary genetic programs, can expand and differentiate to occupy the empty niche and generate the missing organ or cell phenotype of interest. Numerous studies have shown the success of intraspecies and interspecies blastocyst complementation technology for generating liver, forebrain, kidney, pancreas, lung, thyroid, and eye15–20. All organs and cell types have candidate genes that can be targeted to create developmental niches for an organ or cell of interest to be generated via blastocyst complementation 21 . Therefore, blastocyst complementation technology may act as a useful tool for generating cells for repair of the nervous system as well.
Blastocysts Complementation for Repair of the Nervous System
Recent studies have only merely begun to explore blastocyst complementation in nervous system repair. Intraspecies blastocyst complementation in Neurog1+/−-deficient mice produced donor pluripotent stem cell–derived inner ear sensory neurons that successfully corrected inner ear malformations that are typically seen in the heterozygote Neurog1+/−-deficient mice 22 . Another study described neural blastocyst complementation for the generation of forebrain regions. Targeted ablation of host-derived dorsal telencephalic progenitors during development was mediated by Cre-driven diphtheria toxin subunit A targeting Emx1 expressing progenitor cells, resulting in agenesis of the cerebral cortex and hippocampus. Donor mouse embryonic stem cells (ESCs) injected into blastocysts with forebrain-specific ablation allowed for donor stem cells to occupy the developmental niche, generating morphologically and neurologically normal brain tissue 23 . These studies provide evidence that blastocyst complementation can generate healthy nervous system tissue and repair anatomic malformations. However, these studies lack (1) the exploration of specific neuronal cell types that may be generated from blastocyst complementation and (2) how these exogenic neuronal cells may act as a therapy for a multitude of neurodegenerative diseases such as AD. As AD affects a variety of cell types and brain regions, there are many possible types of exogenic cells to consider for potential therapies. Therefore, the following sections review cell types and brain structures affected by neurodegeneration in AD.
Neuropathology and Neurodegeneration in AD
Postmortem examination remains the definitive approach for the clinical diagnosis of AD. The microscopic neuropathology of AD includes a mixed proteinopathy of amyloid beta (Aβ) peptide and tau protein24,25. Aβ and tau accumulate in the brain, preceding the formation of protein oligomers, including amyloid plaques and neurofibrillary tangles (NFTs), respectively24,25. In AD, amyloid plaques and NFTs are commonly distributed throughout the brain in the frontal, temporal, and limbic lobes. Amyloid plaques and NFTs are thought to be neurotoxic and can invoke an inflammatory response of microglia and astrocytes, furthering AD disease progression24,26. Neurodegeneration, the progressive dysfunction and death of neurons, in AD has long been associated with amyloid plaques and NFTs, but explicit causality remains to be determined. These microscopic findings correlate with the macroscopic AD neuropathology of brain atrophy, ventricle enlargement, and volume loss. Typical AD brain atrophy occurs in the frontal, temporal, and limbic lobes. Following atrophy, ventricle enlargement in the frontal and temporal horns of the lateral ventricles is common and volume loss is attributed to both white matter loss and neuronal death24,25. Taken together, these microscopic and macroscopic features of AD create a stereotypic, complex neuropathology that affects many brain regions.
The broad scope of brain regions that may be affected by AD means that disease presentation is likely not identical for each AD patient. Therefore, based on the clinical symptoms and identified brain regions affected in each individual, cellular therapies to supplement and replace specific cell types may be appropriate. A better understanding of the different brain regions and neural cell types affected by AD will provide insight into alternative therapies like exogenic neurons generated by blastocyst complementation.
Hippocampal Formation (Glutamate Pyramidal and Cholinergic Neurons)
The hippocampal formation is part of the limbic and medial temporal lobes and is crucial for normal learning, memory, and emotions27,28. The three main subdivisions of the hippocampal formation are the Cornu Ammonis (CA1, CA2, CA3, and CA4), the dentate gyrus, and the subiculum. The hippocampal formation contains granule cells in the dentate gyrus that give rise to nerve fibers that innervate pyramidal cells from the Cornu Ammonis, as well as glutamate pyramidal neurons29–32. Some important afferent and efferent connections of the hippocampal formation include the basal forebrain, brain stem, cingulate gyrus, and entorhinal cortex. Many studies have detailed neurodegeneration and volume loss in the hippocampus during AD29–32. Hippocampal neurodegeneration in AD patients is believed to initiate within the perforant pathway, a neuronal connection between the entorhinal cortex and hippocampus associated with learning and memory. The Papez circuit, an emotional expression neuronal pathway between the limbic system and hippocampus, is also affected 28 . Amyloid plaques and NFTs accumulate in the transentorhinal cortex before spreading to the entorhinal cortex and hippocampal formation27,28. Disruption of these emotion and learning pathways due to dysfunction and neuronal loss likely contributes to cognitive deficits observed in AD patients28,33.
Several molecular mechanisms have been implicated in volume loss of the hippocampal formation. Amyloid plaques can induce NMDA receptors to degrade postsynaptic glutamatergic receptors at CA1 pyramidal neurons 33 . Cholinergic reorganization, changes in innervation of cholinergic neurons, has also been identified in end-stage AD cases, with increased acetylcholinesterase (AChE) observed in the dentate gyrus 27 . The reorganization of neuronal connectivity is due to amyloid plaques evoking presynaptic release of Ca2+, promoting a positive feedback loop of Aβ aggregation 27 . Over time, there is a progressive loss of the glutamatergic pyramidal neurons and cholinergic medial septal neurons27,33. Cholinergic neuron loss results in decreased cholinergic activity in the hippocampus, leading to further memory deficits and progression of amyloid plaque and NFT pathology in AD34,35. Both reorganization and loss of neurons have been linked to cognitive deficits in AD patients27,28,33. Thus, glutamatergic hippocampal granule and pyramidal cells and cholinergic hippocampal cells should be considered as potential cell populations for generation via blastocyst complementation to treat AD.
Entorhinal Cortex (Glutamate Pyramidal Neurons)
The entorhinal cortex is part of the medial temporal lobe and lies between the transentorhinal area and hippocampal formation. It is associated with memory, navigation, and perception36–40. The entorhinal cortex is composed of six layers and forms connections with several brain regions including the olfactory bulb, subiculum, dentate gyrus, and neocortex, as well as the hippocampus as previously described36–41. Like the hippocampus, the entorhinal cortex contains glutamatergic pyramidal neurons. Notable changes in the entorhinal cortex in AD include atrophy and loss of neuronal and glial cells, as well as glutamate depletion in the perforant pathway32,42–44.
As previously discussed, studies have suggested that amyloid plaques and NFTs originate in the transentorhinal cortex and entorhinal cortex, inducing amyloid plaque and NFT accumulation in the hippocampal formation and cortical structures36–41. Amyloid plaques and NFTs hyperactivate presynaptic and postsynaptic NMDA glutamate receptors in the perforant pathway, which creates an excitotoxic imbalance of glutamate and Ca2+ and decreases synaptic projections37,38,40,41. Amyloid plaques and NFTs also play a role in impairing inhibitory cholinergic and serotonergic synapses that project to the entorhinal cortex, which further contributes to hyperactivity and the propagation of neurodegeneration 38 . Neurodegeneration of the entorhinal cortex is further supported by hallmark AD lesions and pathology studies that result in memory and learning impairments found in AD patients 40 . Thus, entorhinal cortex glutamate pyramidal neurons should also be considered as potential cell populations for generation via blastocyst complementation to treat AD.
Amygdala (Glutamate Pyramidal Neurons)
The amygdala is located in the deep medial temporal lobe and is associated with a variety of functions including emotion, memory, reward, and homeostatic responses45,46. The subnuclei of the amygdala contain many glutamatergic neurons and form connections with the frontal and temporal cortex, olfactory areas, basal forebrain and septal areas, hippocampus, parahippocampus, hypothalamus, thalamus, and brainstem nuclei45,47. AD patients with amygdala neurodegeneration will often present with neuropsychiatric symptoms such as emotional and motivational disturbances, including more agitation/aggression, less irritability, and less anxiety47–49.
Significant neuritic amyloid plaque and NFT pathology in the amygdala are typically present in late AD50–52. In addition, the amygdala is suspected to be the location of secondary misfolding of a-SN and TDP-43 proteins, which are observed in advanced AD 45 . Along with the hallmark histological findings of plaques and tangles, amygdala volume decreases in AD patients compared with healthy controls52–54. The amygdala atrophy observed is comparable with hippocampal atrophy in AD patients and can be detected at various stages of disease severity52,55. Thus, transplantation of exogenic amygdala glutamatergic neurons generated via blastocyst complementation may be considered as a potential AD treatment.
Nucleus Basalis and Medial Septal Nucleus (Cholinergic Neurons)
The basal forebrain is a major cholinergic signaling center located in the front of the brain, deep to the cortex and near the boundary of the frontal and temporal lobes. The main cholinergic signaling center contains structures like the basal ganglia, nucleus accumbens, nucleus basalis, diagonal band, and medial septal nucleus (MSN), and is associated with functions such as sleep and wakefulness, arousal, and learning 56 . Loss and dysfunction of cholinergic neurons, especially in forebrain structures, are commonly seen in AD patients, and some of the first studies which indicated dysfunction of the cholinergic system observed reductions in choline acetyltransferase (ChAT) and ChE activity in the neocortex, nucleus basalis, and MSN57,58. Between 8% and 87% neuronal loss has been reported in the nucleus basalis of AD patients 59 , and lesions in the nucleus basalis and MSN cause AD-like symptoms in mice 60 . The nucleus basalis and MSN project to many brain regions including the amygdala, hippocampus, and entorhinal cortex, which are also affected in AD 61 . These wide-ranging connections likely contribute to the variety of symptoms seen in AD, as nucleus basalis neuronal dysfunction and loss alone could affect signaling in other regions.
The proposed mechanism for basal forebrain cholinergic degeneration begins with hyperphosphorylation of tau protein and the formation of NFTs, leading to neuronal death. Tau pathology and the formation of NFTs, rather than amyloid pathology, are typically associated with basal forebrain–specific degeneration 61 . Potential mediators of neuronal loss include overstimulation via diminishing inhibitory signaling, mitochondrial dysfunction, and chronic inflammation with the release of pro-inflammatory cytokines 62 . Imbalances in trophic factor pathways, such as altered expression of NGF and Trk receptors, and cholinergic receptor have also been implicated in cholinergic neurodegeneration63,64. As cholinergic neurons in the basal forebrain are subject to neurodegeneration and dysfunction in AD, they should be considered as a potential cell population for generation via blastocyst complementation to treat AD.
Locus Coeruleus (Noradrenergic Neurons)
The locus coeruleus (LC) is a pontine nucleus found in the brainstem. LC neurons target hippocampal and cortical regions, including the entorhinal cortex. These connections interface with one another to aid in memory, navigation, and perception 65 . The LC is a primary source of the neurotransmitter norepinephrine/noradrenaline (NE), and NE signaling in the LC is important for autonomic physiological processes—heart rate, sleep and waking patterns, and regulation of neuroinflammation and neuronal survival—and modulates behavior such as attention, stress, and arousal66,67.
The noradrenergic system is affected by AD pathogenesis, leading to neurodegeneration of the LC during disease progression 68 . Significant, progressive neuronal and volume loss in the LC is observed in AD patients68–70. The reduction in neuronal cells is due to AD-specific progression rather than age-related changes 66 . Pre-tangle tau accumulation is detected in the LC neurons’ axons, and in their target neurons, and in later stages of AD, Aβ deposits can be found in the LC71–73. More recent studies have sought to unlock the molecular mechanisms underlying LC neuron involvement. The protein ApoE4, an identified AD risk factor, interacts with vesicular monoamine transporter 2, a transporter responsible for the transport of NE and other neurotransmitters, to inhibit neurotransmitter uptake 74 . NE exclusion from synaptic vesicles leads to oxidation and activation of peptidases within the LC that cleave amyloid plaque and NFT precursors. The cleaved products aggregate and enhance neurotoxicity and degeneration in AD patients74,75. Disruption of mitochondrial and neuroplasticity function has also been implicated in LC neurodegeneration. A ribonucleic acid (RNA) expression analysis of postmortem AD brains found a reduction in respiration genes such as cytochrome C1 and neuron cytoarchitecture genes such as microtubule-associated binding protein 1b 76 . Exactly when these changes occur during AD progression and whether they are related to specific pathology such as NFTs are unclear. However, it is evident that this progressive pathology culminates in LC-NE neuron loss.
Maintaining NE signaling protects LC-NE neurons, which safeguards LC-specific and noradrenergic system functions: arousal, mental challenge, and novel situations 77 . Recent evidence demonstrates that maintaining neural density of LC-NE neurons prevents cognitive decline in aging 78 . As maintenance of LC-NE neurons has a neuroprotective role and noradrenergic neuron dysfunction influences AD progression, the use of exogenic LC-NE neurons generated via blastocyst complementation should be considered as a potential avenue to treat AD.
Dorsal Raphe (Serotonergic Neurons)
The dorsal raphe nucleus (DRN) is a structure in the bilateral midbrain containing approximately 235,000 neurons, roughly 70% of which utilize the neurotransmitter serotonin79,80. The DRN projects efferent serotonergic neurons in a topographically organized manner along the rostrocaudal axis 81 . Brain regions targeted by the DRN include the cerebral, transentorhinal, entorhinal, and prefrontal cortex; hippocampal formation; olfactory bulb; striatum; septum; amygdala; some thalamic nuclei; and several brainstem nuclei 82 . Due to the vast number of connections to other brain regions, the DRN may act as a modulator of complex autonomic functions 81 .
Cellular and morphological alterations in the DRN can be caused by AD. Significant cell loss is observed in AD patients, which is characterized by decreased cell packing density and the increased proportion of large neurons present70,83. Besides cell loss, remaining DRN neurons have diminished serotonin levels 84 . Decreased serotonergic signaling is also observed in the amygdala, striatum, and substantia nigra of AD patients 65 . Tryptophan hydroxylase (TPH), the principal enzyme required to synthesize serotonin, shows reduced activity in the raphe nuclei cells in AD85,86. DRN neurodegeneration is seen in multiple mouse AD models and is linked to atypical, ectopic cell cycle events in postmitotic neurons 87 . In addition to the cellular changes, gross hallmark morphological changes occur in the DRN of the AD brain. The DRN is among the first brain regions affected by cytoskeletal changes associated with NFTs in AD, with NFTs first appearing in the DRN in early stages of AD81,83,88,89. Taken together, the cellular and morphological changes suggest that neurodegeneration of the DRN affects AD progression and pathology. Thus, treatment of AD with exogenic serotonergic neurons of the DRN generated via blastocyst complementation should be considered.
Interneurons
Interneurons are a heterogeneous group of neurons with diverse morphologies and biochemical and physiological properties. They are found throughout the brain, notably in the cortex, and form complex connections that modulate neural circuits via inhibitory and excitatory signaling. Inhibitory interneurons account for approximately 20% of all cortical neurons, and use gamma-aminobutyric acid (GABA) as their neurotransmitter 90 . Interneuron morphology is variable and is influenced by the shape and orientation of neighboring dendrites and axons. Based on their morphology and the expression of specific molecular markers, interneurons can be classified into subtypes, like parvalbumin (PV) expressing chandelier and multipolar basket cells, somatostatin (SST) expressing Martinotti cells, and others which express neuropeptide Y (NPY), vasoactive intestinal peptide (VIP), and cholecystokinin (CCK) 91 . The molecular markers influence the function of each subtype including their connectivity and synaptic activity 92 . Interneurons can also be classified by their electrophysiological properties, which include fast-spiking interneurons, non–fast-spiking interneurons, adapting interneurons, irregular spiking interneurons, intrinsic bursting interneurons, and accelerating interneurons91,93. A variety of ionic channels expressed intrinsically in interneurons establishes their unique electrophysiology and function 94 . The combination of these diverse properties allows interneurons to establish synapses with different subcellular regions of their neuronal targets 94 .
GABAergic interneuron dysfunction can occur in AD, and the resulting imbalance between excitation and inhibition within the brain leads to network hyperexcitability and affects neurodegenerative processes90,95. In a meta-analysis of AD models and interneurons, Reid et al. found that most hippocampal interneurons expressing CCK were not affected by AD until old age, while NPY interneurons express reduced levels of NPY in the presence of AD pathology96,97. PV interneurons had reduced levels of PV expression in AD, as early as 1 month after pathology onset 97 . In addition, dysfunction of fast-spiking PV interneurons, a source of inhibition in the cerebral cortex, has been specifically associated with abnormal gamma activity, oscillatory firing of neurons in the frequency of 30–120 Hz, in the hAPP AD mouse model 98 . Both AD humans and animal models also show decreased SST expression96,99–101. Thus, a variety of interneuron subtypes may become dysfunctional in AD.
At a molecular level, interneurons are affected by Aβ plaques and tau accumulation, leading to hyperexcitability of neural networks. One consequence of hyperexcitability is that AD patients experience higher incidences of seizures 102 . Increased Aβ deposition is associated with excitation of cortical and hippocampal networks that results in spontaneous nonconvulsive seizures 103 . The interneurons in these hyperactive neuronal networks of AD hAPP/PS1 mice showed decreased GABAergic inhibition 104 . GABAergic interneurons may be more vulnerable to AD pathology in certain AD mouse models such as TgCRND8 and TauPS2APP105,106. Tau accumulation in GABAergic interneurons was observed in both 3xTg mice and AD patients 107 . ApoE4-KI mouse models have shown mixed results, with some ApoE genotypes decreasing the number of GABAergic interneurons and their function in the hippocampus, specifically the dentate gyrus, while others show no effect on GABAergic interneurons108–112. ApoE4 mice generate more neurotoxic fragments than wild-type mice, leading to increased tau phosphorylation that worsens AD neuropathology108,110,113. Other AD mouse models such as APP/PS1 mice show progressive loss of GAD67+, Calretinin+, and NPY+, and PV+ interneurons in the hippocampus as early as 7 months of age, which is closely followed by cognitive deficits and AD-like pathology 114 . In summary, the neuronal hyperexcitability observed in AD patients is also driven by a decrease in GABAergic interneuron inhibition that leads to memory and cognitive impairments 115 . Thus, various types of interneurons should be considered as potential cell populations for generation via blastocyst complementation to treat AD.
Preclinical Transplantation Studies in Experimental Models of AD
Historically, the investigation of AD and its possible therapeutics such as transplantation have been studied using transgenic AD animal models that were generated by targeting AD risk genes associated with early onset AD (eg, APP, PSEN1, and PSEN2) and late onset AD (eg, APOE4)116,117. These transgenic animal models have included mouse, rat, primate, pig, sheep, and canine models of AD116–120. The various AD models at the disposal of the scientific community enable transplantation studies with multiple neuronal types that may alleviate or rescue AD symptoms and possibly alter AD pathogenesis. Here, we have reviewed transplantation studies to provide further insight into potential cell types that may be derived from blastocyst complementation to treat AD.
Neural Cell Phenotypes for Transplantation in Experimental Models of AD Hippocampal Neurons
Hippocampal neurodegeneration in AD involves an aberrant change in neuron organization that induces excitotoxicity, loss of neurotrophic factors, and damage to neural circuitry, ultimately leading to a decline in learning and memory 121 . Hippocampal neuron transplantation provides a potential therapeutic approach for normalization of neurotrophic levels and functional recovery of neural circuits121,122. In rat models with hippocampal neurodegeneration and cognitive impairment, transplanted fetal rat hippocampal neurons expressed brain-derived neurotrophic factor (BDNF) and basic fibroblast growth factor (FGF)121–123. Neurotrophic factor expression was normalized in aged rats after hippocampal neuron transplants enriched with FGF-2123. Additional pathologies are corrected after hippocampal transplantation, which include excitotoxicity, aberrant growth of mossy fibers, and abnormal levels of GABA and calcium-binding protein calbindin123–126. These factors generate a supportive microenvironment, suggesting that hippocampal transplants can improve the survival of host neurons and potentially lead to improvements in cognitive impairments in AD121,122. Together these studies demonstrate that hippocampal neuron transplantation can provide neurotrophic support and promote functional recovery of cognitive impairment and alleviate dementia found in AD. This is further evidence that hippocampal neurons are a potential cell population, which could be generated by blastocyst complementation for the treatment of AD.
Cholinergic Neurons
A significant loss of both cholinergic cells and neural activity is observed in the basal forebrain and hippocampus of AD patients and is associated with common behavioral deficits, especially memory loss 127 . Cholinergic neuron transplantation may be a potential therapeutic strategy to rescue cholinergic neuron loss, restore cholinergic signaling, and reverse cognitive deficits seen in AD. Several studies conducted in the 1980s and 1990s investigated the transplantation of primary embryonic cells. Cholinergic neurons derived from the MSN of embryonic-day-15 rat embryos were transplanted into the hippocampal formation of adult rats with MSN-hippocampal pathway lesions that resulted in the reinnervation of the hippocampal pyramidal cell neuronal layers and the granule cell layer of the dentate gyrus 128 . Moreover, reinstatement of this neural connectivity restored spatial learning and memory function 129 . Rat cholinergic neurons extracted from embryonic days 14 and 15 were transplanted into aged rat’s hippocampi and successfully engrafted, expressed AChE, and grafted animals displayed marked improvements in memory and behavior tasks with no obvious impairments 130 . In a primate study, transplantation of fetal marmoset basal forebrain tissue resulted in AChE expression and moderate improvements in learning and memory tasks in marmosets with chemical lesions in the nucleus basalis 131 . Another approach examined was autotransplantation, the transplantation of organs or tissues from one region of the body to another in the same individual. Nodose ganglion cells autotransplanted into the right parietal cortex of rats with chemically induced lesions in the nucleus basalis survived and expressed AChE, leading to mild improvements in behavior and memory retention of autotransplanted rats compared with sham animals 132 . These studies suggest that cholinergic neuron transplantation can successfully rescue loss of cholinergic signaling and reverse cognitive impairments.
These early strategies for cholinergic neural transplantation showed promising results. However, their translational value is limited by ethical concerns regarding the use of primary fetal tissue and autotransplantation as well as practical challenges such as scalability and homogeneity of the product. Therefore, recent work has focused on the use of stem cells and other stem cell–derived cholinergic neurons. Several differentiation protocols have been published for the conversion of pluripotent cells into cholinergic neurons with over 80% purity133,134. These protocols rely on supplementation of morphogens or transcription factors such as growth factors and bone morphogenetic proteins.
Transplantation experiments have reported using both mouse and human stem cell–derived cholinergic cells. Human iPSC-derived cholinergic neurons transplanted into the bilateral hippocampus of an aged AD mouse model, PDAPP that overexpress a mutant human amyloid precursor proteins, expressed ChAT and survived up to 45 days post-transplantation 135 . Mouse neural stem cell–derived cholinergic neurons transplanted into the hippocampus of APP/PS1 mice survived 3 months post-transplantation, expressed ChAT in approximately 72.6% of the cells, and formed functional synapses with host tissue 134 . Both human and mouse ESC-derived basal forebrain cholinergic cells grafted into the nucleus basalis of 5XFAD and APP/PS1 mice survived 2 months post-transplantation, expressed ChAT, and showed electrophysiological characteristics similar to mature cholinergic neurons 136 . In this study, human ESC-derived cells had diminished transplantation success compared with mouse ESC-derived cells, likely due to species-specific differences.
Both mouse neural stem cell and ESC-derived cholinergic grafts transplanted into AD mice ameliorated neuropathological disturbances like swollen mitochondria, synapse loss, necrosis of axons, and induced secretion of neurotrophic factors134,136. However, no effect was observed on the total number of Aβ plaques134,136. The transplanted AD mice also showed improvements in memory tasks compared with non-transplanted AD mice134–136. These studies suggest successful integration of transplanted cells into endogenous neural circuitry and correction of cholinergic deficits typically observed in these AD mouse models. Thus, cholinergic neurons should also be considered as a potential cell type to be generated using blastocyst complementation for the treatment of AD.
Noradrenergic Neurons
Noradrenergic deficits occur in AD, especially in the LC, and transplantation of NE-expressing cells has been explored as a treatment for AD to restore these deficits. Fetal rat LC noradrenergic neurons transplanted into the third cerebral ventricle normalized retention performance of inhibitory avoidance tasks in aged rats. The noradrenergic neuron grafts contained fluorescent NE-expressing neurons with extensive networks, which demonstrated fusion between graft and host brain at one or more zones along the third ventricle walls, with some noradrenergic fibers crossing the graft–host interfaces 137 . Noradrenergic fetal rat grafts also partially normalized noradrenergic hippocampal regions depleted by chemical lesions and delayed seizure activity, restored NE synthesis rate close to wild-type animals, and reinnervated the region with a 40% survival rate138,139. Allotransplantation of embryonic LC grafts also restored normal behavior after chemical lesions of the endogenous noradrenergic system 140 . Additional studies demonstrated that LC noradrenergic neurons from fetal rats transplanted to a denervated lumbar spinal cord can restore NE levels up to 60% of the controls 141 . Although these studies occurred several decades ago, they provide insight into LC neurons’ ability to restore noradrenergic systems in other injured central nervous system regions. According to these studies, transplanted noradrenergic neurons can engraft and synthesize NE. These studies suggest that noradrenergic neurons may provide a successful cell type for transplantation to recover behavioral and anatomical deficits observed in AD patients, and thus the generation of NE neurons by blastocyst complementation should be considered.
Serotonergic Neurons
Serotonergic neuron loss leads to cognitive deficits including memory processes, spatial navigation, decision-making, and social relationships 142 . Numerous studies have explored the transplantation of serotonergic neurons, and their ability to alleviate and partially rescue cognitive symptoms associated with serotonergic neuron loss. Due to serotonergic neuron loss observed in AD, this may prove to be a promising cell type for transplantation.
Fetal rat serotonergic neurons have been grafted to an adult rat hippocampus and spinal cord, and these grafted neurons survive, innervate, and may restore the neurotransmitter 5-HT143–146. Serotonergic neurons from fetal rats grafted into the dentate gyrus of adult rats with chemically lesioned hippocampi can reinnervate the region with dense populations of serotonergic neurons derived from the transplanted cells147,148. Specifically, serotonergic neurons taken from the dorsal raphe can reverse the 5-HT depletion induced by the hyperexcitability of granule cells and partly regulate granule cell proliferation in the dentate gyrus148,149. Fetal serotonergic raphe grafts can lead to compensation or overcompensation of 5-HT that results in accumulation of 5-HT, evoking further release of 5-HT from the hippocampal regions150,151. In addition, implanting serotonergic neuron-enriched embryonic raphe nucleus-derived rat neural stem cells into spinal cord injuries of female rats led to serotonergic axons projecting to autonomic regions such as autonomic circuits associated with the kidney and cardiovascular system 152 . These studies suggest that the raphe is a serotonergic neuron-rich brain region that acts as a plausible source for donor graft tissue and can provide functional neurons to both the brain and spinal cord.
The mixed transplantation of embryonic rat serotonergic and cholinergic neurons has shown improvements in spatial memory tasks in rats 10 months post-transplantation compared with transplanted rats with one graft type, which were unable to rescue the combined denervation of cholinergic and serotonergic neurons 143 . Another study found that serotonergic neuron-rich grafts from rats will overcompensate chemical lesion–induced serotonergic deficits in adult rats, while a co-grafting technique with cholinergic neurons will stop cholinergic deficits and limit the overcompensation of the serotonergic deficits, ultimately leading to recovery of both serotonergic and cholinergic deficits 153 . These studies show that there is a partial cognitive recovery after serotonergic grafts, and that serotonergic transplants may alleviate cognitive impairments due to AD pathology associated with the serotonergic systems. Therefore, the generation of serotonergic neurons via blastocyst complementation is a strategy, which should be investigated.
Interneurons
As AD progresses, various interneuron subtypes (eg, PV+ and SST+ cells) become dysfunctional or are lost, resulting in an imbalance of excitatory/inhibitory signaling that is correlated with cognitive deficits98,99. Thus, the transplantation of healthy interneurons to supplement and replace damaged tissue has been explored as a therapeutic AD intervention.
Fetal medial ganglionic eminence (MGE)–derived progenitor cells have shown promise as a transplantation strategy. The MGE is a major source of interneurons; it is estimated that 60–70% of cortical interneurons are derived from this region154–156. Numerous studies have interrogated MGE cell transplantation and found that these cells are capable of long-term engraftment, survival, differentiation into interneuron subtypes, formation of synaptic connections, and migration156–159. However, the exact mechanisms of MGE engraftment and differentiation are still under investigation. Two commonly proposed theories of MGE engraftment are (1) MGE cells permit the “re-opening” of a transient window of plasticity from early development and (2) transplanted cells “rejuvenate” host cells via the release of beneficial trophic factors160,161. Evidence for these theories comes from studies in which transplantation of MGE cells has induced rewiring leading to changes in ocular dominance in mice160,161. The visual cortex plasticity observed after MGE cell transplants can be mediated by neuregulin/ErbB4 signaling in host interneurons 107 . Thus, both neural network rewriting and the secretion of beneficial trophic factors act as potential mechanisms by which transplanted interneurons improve host brain function.
The plasticity of MGE cells has been further addressed with transplantation studies aimed to understand how age of the donor cells and recipient affects the success of transplantation. MGE cells were isolated and transplanted into different aged mouse embryos to investigate if they contribute to different cortical regions in the recipient animal. “Early” progenitors contributed to lower cortical layers, while “late” progenitors contributed to upper layers in age-matched mice recipients. “Fate-switching” was observed when different aged progenitor cells were transplanted into different age recipients, which led to contributions to all layers of the cortex in heterochronic transplants 162 . These results indicate that MGE-sourced cells retain a level of plasticity, and their cell fate is influenced by extrinsic factors in the host brain. This study suggests that transplanted progenitor cells will integrate appropriately to the host tissue organization.
MGE cell transplantation has been studied in various disease models including seizure disorders, Parkinson’s disease, neuropathic pain, stroke, anxiety, psychosis, and AD163–166. Several studies have shown that MGE cells from fetal mice transplanted into the hippocampi of AD mice survive and integrate into the host tissue for over 3 months, and can generate interneuron subtypes (eg, SST+, NPY+, and PV+) that have electrophysiological properties comparable with those of endogenous interneurons114,165. However, one study found that the number of transplanted cells progressively decreased, with approximately 15% remaining at 60 days post-transplantation 114 . Abnormal increases in neural activity including “epileptic-like” spikes, particularly in the hippocampus, are typically observed in AD models 167 . Thus, MGE transplantation studies have assessed the electrophysiological properties of transplanted cells to determine the effect of MGE transplants on excitatory–inhibitory signaling160,161. Engineering MGE cells prior to transplantation may optimize their therapeutic effect. Overexpression of the sodium channel Nav1.1 in MGE transplants in APP J20 mice improved cognition, normalized electrophysiological abnormalities, including network hypersynchrony, and altered the amount of synchronous activity 168 . These results suggest genetic engineering as an additional dimension to MGE transplantation therapy.
In another study, ApoE4-KI mice that received MGE transplant interneurons exhibited increased spontaneous inhibitory postsynaptic potentials in the dentate gyri, implying functional alterations of neural circuits 165 . A separate study tested MGE transplants into an APP/PS1 mouse model, and showed a decrease in the number of epileptic spikes and had improvements in long-term potentiation compared with non-transplanted APP/PS1 mice 114 . In both studies, transplanted AD mouse models showed improvements in learning and memory tasks114,165. However, amyloid accumulation in FAD mice has been linked to disruption of neural circuitry 98 . However, MGE transplantation did not appear to affect amyloid burden 165 . Despite the continued presence of amyloid, the survival of MGE grafts suggests that donor cells are not immediately susceptible to toxic amyloid host pathology and may retain their efficacy over time, avoiding the need for repeat treatments.
A step toward clinical application of MGE transplants is the transplantation of human MGE cells into mouse models. When human ESC-derived MGE-like cells were transplanted into the hippocampus of a chemically induced learning and memory deficits mouse model, the transplanted cells engrafted and migrated throughout the brain. Minor anatomical alterations were observed, including enlarged regions near the injection site and in the dentate gyrus 169 . Most of the transplanted cells, approximately 45%, were GABA+ at 6 months post-transplantation, and various interneuron subtypes were observed among the transplanted cells, including SST+, PV+, calbindin, and NPY+ cells 169 . Human ESC-derived MGE-like cell transplanted mice had improved performance in learning and memory tasks compared with non-transplanted mice 169 . This study suggests that human cells provide cognitive improvements with restoration of some interneurons. Taken together, these studies provide evidence of physiological and functional improvement in AD mouse models and memory loss after interneuron transplantation, indicating that interneurons should be considered as a potential AD therapeutic to be generated via blastocyst complementation.
Developmental Neurobiology as a Guide for Identifying Targets for Gene Editing and the Generation of Exogenic Neurons
The preceding sections reviewed areas of the brain affected by AD and previous transplantation studies to identify cell populations of interest, which could be generated through blastocyst complementation for the treatment of AD. A critical step in generating these exogenic cells via blastocyst complementation is the identification of key genes involved in the specification and development of the target cell of interest. The following is a review of the developmental neurobiology of candidate neuronal cells for generating exogenic neurons for treating AD and the identification of key genes for gene editing. Particular attention has been made to knockout studies, as the extent, specificity, and timing of neuronal agenesis implies the success to which ablating that gene will generate a developmental niche to be filled by donor cells.
Developmental Neurobiology of the Hippocampal Neurons and Limbic Projection Neurons
The hippocampal formation contains glutamatergic pyramidal-projection cells in the CA and granule cells in the dentate gyrus. These hippocampal neurons arise from the cortical hem (CH) at approximately E10.5, and then further develop into the choroid plexus (ChP), Cajal–Retzius (CR) cells, and hippocampus 170 . These three regions construct the primitive hippocampal formation. Each region of the primitive hippocampal formation gives rise to different progenitors and cells types, which are as follows: (1) the hippocampus gives rise to pyramidal cells that project to various cortical regions, (2) CR cells give rise to the early neuronal progenitors, (3) CH and CR cells give rise to dentate gyrus progenitor cells, and (4) the ChP gives rise to neural stem cells (Fig. 1A)170–173.

Development of glutamatergic neurons in the hippocampus and limbic projection neurons with their associated transcription factors and morphogens. (A) Glutamatergic pyramidal-projection neurons develop from CH, which further develop into the hippocampus, ChP, and CR cells. (B) The induction and inhibition of several transcription factors (black text), morphogens (orange-brown boxes), and cellular signals guide the development of glutamatergic pyramidal-projection neurons and granule cells. CH: cortical hem; ChP: choroid plexus; CR: Cajal–Retzius.
Important transcription factors and morphogens can be identified as developmental regulators of hippocampal progenitor neurons. Known signaling pathways such as bone morphogenetic protein (BMP), Wingless/Integrated (Wnt), FGFs, sonic hedgehog (Shh), and Reelin regulate the growth of hippocampal progenitors in the dorsal and medial telencephalon (Fig. 1B)170–174. More specifically, BMP signaling and Wnt signaling induce dorsal growth of the CH, while FGF signaling and Shh signaling regulate the growth of the CH by inducing ventralization of the telencephalon170,174. Further development of the CH into the ChP at approximately E11.5 is affected by downstream homeobox transcription factors, while CR cell migration guidance is instructed by Reelin expression170,174.
The main inductive morphogens of the dorsal telencephalon into the CH are BMP and Wnt. Wnt3a inactivation severely reduced hippocampal tissue 175 . Diminished Wnt3a function resulted in decreased hippocampal formation, while maintaining dentate gyrus progenitor cells171,175. Further growth and differentiation of the CH into the hippocampus and ChP are regulated by Emx1/Emx2, Otx1/Otx2, Lef1, Gli3, Foxg1, Pax6, and Lhx2 (Fig. 1B)170,173–179. Mutation and knockout studies of transcription factors Emx1/Emx2, Otx1/Otx2, Lef1, and Gli3 have shown loss of the hippocampus and dentate gyrus formation, indicating their role in hippocampal formation170,174–176,179. Emx1/Emx2 and Otx1/Otx2 knockout studies combined with transcriptional analysis showed that Wnt signaling and BMP signaling regulate these key hippocampus development transcription factors170,175. Gli3 is an upstream transcription factor of Wnt and has positive feedback on Wnt signaling and Emx1/Emx2 expression 175 . These transcriptional effectors cooperate to regulate dorsalization and growth of the CH23,170,175,179.
Additional mutation and knockout studies of Foxg1, Pax6, and Lhx2 have shown abnormal growth of the hippocampus due to decreased lateralization and ventralization of the telencephalon170,173,174,177,178. Lhx2 is a central regulator of neuron-glia cell fate in the hippocampal formation, as it mediates dorsal and ventral patterning of the telencephalon170,173,177,178. Knocking out Lhx2 results in a larger CH that extends into the ventral telencephalon, while overexpression of Lhx2 leads to inhibition of Notch-induced astrogliogenesis and prolonged neurogenesis 173 . Lhx2 works cooperatively with Pax6 to regulate the CH by controlling the medial position of the hippocampal formation 177 . Double mutation of Lhx2/Pax6 results in a larger CH that extends to the ventral telencephalon due to loss of Lhx2 and to the lateral regions of the telencephalon due to the loss of Pax6 177 . Foxg1 is a regulator of Lhx2 expression and promotes ventral patterning of the hippocampal formation 178 . Foxg1 operates independently of Shh; however, Shh knockout impairs Foxg1 function, demonstrating a cooperative function with Shh174,177,178. Foxg1 also cooperates with FGFs in the ventralization of the telencephalon 174 . Taken together, these studies show that each of these genes plays a key role in normal hippocampal development and that by targeting these transcription factors, hippocampal neurons may be generated through blastocyst complementation.
At E12.5, development of CR cells and dentate gyrus progenitors follows different signaling pathways that include Reelin and Lef1/TCF family of transcription factors. CR cells develop from the CH, which is driven by the expression of Reelin, a glycoprotein that is critical for neuronal migration, and p73, a transcription regulator that is essential for immature neural progenitor cells and survival of postmitotic neurons170,172,180. Knocking out p73 leads to a loss of CR cells and CH patterning in the amygdala, dentate gyrus, and hippocampus172,180. Studies of the dentate gyrus development have shown that the Lef1/TCF family, a nuclear mediator of Wnt signaling, plays a role in hippocampal formation. By knocking out Lef1, the development of the dentate gyrus granule cells is restricted, while a double knockout of Lef1/Tcf1 lacked the entire hippocampus170,175,176. These findings suggest that these transcription factors are target candidates for the CR cells and dentate gyrus progenitors found within the hippocampal formation.
To target glutamatergic pyramidal-projection neurons, the transcriptional factors that regulate neurogenesis in the limbic cortex and hippocampal formation must be discussed. The glutamatergic pyramidal-projection neurons develop around E14.5170,181. Neurogenesis of pyramidal-projection neurons occurs in the embryonic neocortex and subgranular zone of the dentate gyrus181,182. Early progenitor radial-glial cells express Pax6, and then express Tbr2 as early intermediate progenitor neurons and NeuroD as late intermediate progenitor neurons181,182. Pax6 is important in glutamatergic neuron, GABAergic neuron, and astrocyte development181,182. Tbr2 and NeuroD have been implicated in glutamatergic neuron proliferation, differentiation, and migration181,182. Mature glutamatergic pyramidal-projection neurons express Tbr1181,182. Tbr1 expression is important for layer-related differentiation, migration, and axon projections181,182. These transcription factors, Pax6, Tbr2, NeuroD, and Tbr1, are expressed sequentially during neurogenesis of glutamatergic pyramidal-projection neurons 181 . By targeting these transcription factors, glutamatergic pyramidal-projection neurons from the hippocampal formation and limbic cortex may be generated from blastocyst complementation.
Developmental Biology of Cholinergic Nucleus Basalis and MSN Neurons
Cholinergic neurons of the basal forebrain, especially the nucleus basalis of Meynert (NBM) and MSN, are commonly affected in AD. To identify potential target genes for knockout and the generation of exogenic cholinergic neurons using blastocyst complementation, an understanding of cholinergic developmental biology is needed.
A significant population of cholinergic neurons originate from Nkx2.1+ progenitor cells in the subpallium/ventral telencephalon 183 . Expression of Nkx2.1 occurs between E9.5 and E10.5 in the MGE, septum, and preoptic area (POA; Fig. 2A)184,185. Knocking out Nkx2.1 results in a significant loss of cholinergic cells throughout the basal forebrain 186 . Nkx2.1 expression in cholinergic cells is maintained into adulthood, and postnatal knockout is associated with degeneration of cholinergic cells, suggesting Nkx2.1 expression is vital at all stages of cholinergic development183,187. Olig2 has also been associated with early (E9 and E10) cholinergic specification, and loss of Olig2 led to a slight reduction in ChAT+ cells 188 . Further patterning of cholinergic neuron subtypes is regulated by additional transcription factors. Lhx8, historically referred to as Lhx7, is expressed around E10.5 and found downstream of Nkx2 183 . Lhx8 is strongly associated with differentiation and specification of cholinergic neurons189,190. Lhx8 KO mice exhibit severe loss of cholinergic neurons in the caudate putamen and nucleus basalis183,191. Binding partners of Lhx8 are also implicated in cholinergic differentiation, including Isl1 and Ldb1183,184. Lhx8 and Isl1 form a complex that binds cholinergic enhancer sequences like ChAT190,192. Conditional KO of Isl1 led to a partial reduction in cholinergic neurons190,192. Similarly, conditional KO of Lbd1 in Nkx2.1 lineage cells led to a reduction in ChAT+ cells 193 . The transcription factors Gbx1 and Gbx2 further regulate cholinergic development downstream of Lhx8183,184. Gbx1 is expressed with Lhx8 in basal forebrain cholinergic nuclei, and overexpression promotes cholinergic fate of ESCs in vitro133,194. Loss of cholinergic neurons, specifically late-born striatal interneurons, was reported upon KO of Gbx2 195 . Some studies have also implicated Otx2 in late cholinergic specification190,196. A population of forebrain cholinergic neurons which originate from the ventral pallium, rather than the subpallium, have also been identified. These cells express Tbr1 and contribute to the horizontal diagonal band, nucleus basalis, and amygdala (Fig. 2B, C) 197 .
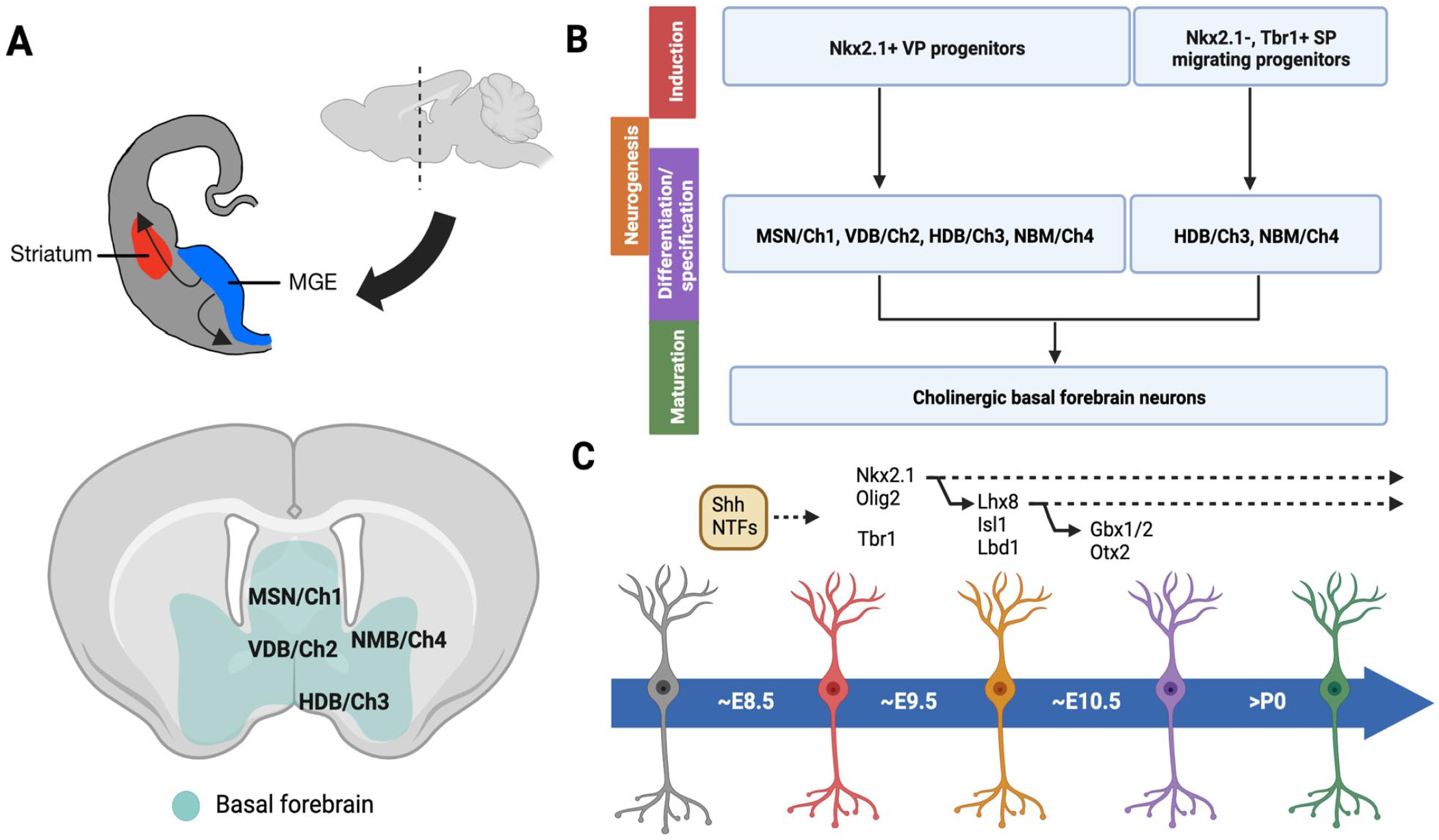
Development of cholinergic nucleus basalis and medial septal nucleus neurons with their associated transcription factors and morphogens. (A) Embryonic origin of cholinergic progenitor cells. Nkx2.1+ cells originate in the MGE (blue) and migrate laterally throughout the cortex (gray) and striatum (red). Basal forebrain (green) structures in the adult mouse are indicated, including the MSN/Ch1, VDB/Ch2, HDB/Ch3, and NBM/Ch4. (B) Separate origins and contributions of Nkx2.1+ and Nkx2.1− Tbr+ cholinergic progenitors. Stages of development include induction (red), neurogenesis (orange), differentiation/specification (purple), and maturation (green). (C) Approximate timeline of important transcription factors (black text), morphogens, and NTF (orange-brown box) expressed during development of cholinergic basal forebrain neurons. MGE: medial ganglionic eminence; MSN: medial septal nucleus; VDB: vertical diagonal band; HDB: horizontal diagonal band; NBM: nucleus basalis of Meynert; NTF: neurotrophic factors.
Besides transcription factors, morphogen signaling also influences cholinergic neuron development. Shh is known to drive Nkx2.1 expression around E9.5 183 . Neurotrophic factors such as NGF, FGFs, BMP9, and BDNF are involved in differentiation, maturation, and migration of cholinergic cells, yet their role in early specification is unknown. Cholinergic cells express neurotrophin receptors TrkA at E18 and p75NTR at E11 and neurotrophic factor NGF, which has a positive feedback loop with Lhx8183,185,198. Cholinergic progenitors that later contribute to the NBM and MSN expressed Fgf8 and Fgf17 as early as E8.5 199 . However, the KO or disruption of neurotrophins has variable effects on cholinergic neuron development, such as varied changes in cell size, number, and connectivity185,200–205. The role of p75NTR is complex. Knocking out p75NTR increased basal forebrain cholinergic neurons in adult animals, while perinatal and postnatal conditional knockouts reduced cholinergic neurons 185 . These findings suggest each neurotrophic factor is not individually necessary, but is still important for appropriate cholinergic development, especially for maturation in later stages of development. In summary, targeting key transcription factors including Nk2.1, Lhx8, and Isl1 and/or neurotrophic factor pathways such as NGF may enable the generation of forebrain cholinergic neurons using blastocyst complementation.
Developmental Neurobiology of Noradrenergic LC Neurons
The LC contains several neuron types with varying morphologies and chemical characteristics. However, the most prominent neuron type is the NE-synthesizing cells. The neurons located in the LC that release NE are immunoreactive for the enzymes tyrosine hydroxylase and dopamine-B-hydroxylase (DBH), which are critical for NE biosynthesis 206 . LC neurons may also express NPY, SST, CCK, galanin, and adrenoceptor207,208. As the LC is one of the primary brain regions for NE synthesis and is one of the first locations vulnerable in AD, it is important to consider the neurobiology and development of the LC and NE neurons for the potential generation of exogenic LC neurons for transplantation.
The LC progenitor cells originate in the rostral rhombic lip and migrate ventrally within the neural tube until they stop their migration and reside in the pons near the fourth ventricle 209 . Development of LC noradrenergic neurons is determined by several molecular mechanisms. The Phox2 gene is expressed in all neuronal types that transiently or permanently express DBH that is needed for NE biosynthesis 210 . Phox2a loss of function results in partial agenesis of the LC, and when Phox2b is inactive the LC progenitor cells will not differentiate into noradrenergic neurons211,212. Phox2 gene expression is further driven by Mash1, also known as Ascl1, which is found upstream and expressed as early as E9.5 in the mouse213,214. Additional transcription factors and proteins that participate in noradrenergic differentiation and formation of the LC include: Rnx, BMPs, Pax3, Pax6, Pax7, FGFs, and orphan receptors Ear2 (Nr2f6)210,215–218.
Phox2 expression is an early marker for noradrenergic neurons and gives insight into LC generation and development. Of the transcription factors and proteins mentioned above, there are several that show further roles in LC formation. Phox2 expression near Bmp5-expressing cells in the dorsal neural folds and roof plate influences LC development. Notably, when BMPs are inhibited at embryonic stage 10 (33–45 h) in chick embryos by Noggin, the inhibition will prevent the generation of LC neurons or lead to a dorsal midline localization of the LC neurons217,219. The dorsal BMP signaling centers affect the roof plate and rhombic lip formation, which is affected when inhibiting BMPs. While at stage 13/14 (50–52 h) of chick embryo development, embryos co-express Phox2, Pax3, and Pax6 217 . Pax3 and Pax6 play a role in dorsoventral patterning and thus are observed in a more ventrolateral position in association with Phox2 around stage 5 of chick development211,217,220. Pax3, Pax6, and Pax7 are observed much earlier in the mouse at approximately E8.0–E8.5 and are expressed throughout the development of the central nervous system (Fig. 3) 221 . The described molecular factors important for LC and noradrenergic neuron development provide candidates for generating exogenic neurons in the LC and more specifically the noradrenergic neurons that are affected by AD.

Development of LC with their associated transcription factors and morphogens in the chick and mouse embryo. The chick embryo develops over 21 days with 46 stages, while the mouse ranges from 19 to 21 days with 28 stages. Each stage for the two species has key developmental and transcriptomic architecture. Based primarily on the transcriptomic architecture for each species, the chick’s 33–45 h (stage 10, 10–15 somites) development is similar to that of embryonic day 9 (stage 14, 13–20 somites) in a mouse, and the chick’s 50–52 h (stage 13/14, 20–21 somites) development is similar to that of embryonic day 13 (stage 21, 52–55 somites) in the mouse. Key transcripts (black text) and morphogens (orange-brown boxes) are depicted for the chick (top) and the mouse (bottom) with their approximate expression throughout each species’ development. LC: locus coeruleus.
Developmental Neurobiology of Serotonergic Raphe Neurons
The raphe nuclei contain heterogeneous population of neurons with distinct morphologies, projections, and neurochemical characteristics. Serotonergic neurons are one of the major neuron types present in the raphe nuclei 222 . The serotonergic neurons are known to synthesize serotonin through the presence of serotonin itself or the biosynthetic enzyme, TPH223,224. The early presence of the serotonergic system is critical for the synthesis of serotonin and growth of axons in the developing raphe neurons, as these cells undergo cell division, migration, and differentiation during development. These developmental mechanisms in the brain may be modulated by serotonin to some extent 225 . As the raphe nuclei are rich in serotonergic neurons and the dorsal raphe is susceptible to AD pathology, it is important to consider the neurobiology and development of the raphe nuclei and the serotonergic neurons.
The raphe population is divided into two main clusters: (1) a rostral cluster found in the pons and (2) a caudal cluster found in the medulla 226 . The two clusters found during embryogenesis contain several compartments of the hindbrain called rhombomeres (Fig. 4A) 227 . The rostral cluster contains rhombomeres 1–3 and the caudal cluster contains rhombomeres 5–7. The rostral cluster contains the dorsal raphe and median raphe nuclei 226 . More specifically, rhombomere 1 gives rise to the DRNs and rhombomeres 2 and 3 give rise to the median raphe nuclei (Fig. 4B) 228 . All the neurons found in the raphe nuclei express TPH for serotonin synthesis, yet all cells do not actively produce serotonin, meaning they operate under polymorphisms associated with functional changes determined by protein levels or bioactivity of the proteins 229 . Early embryogenesis determines the neural axes as dorsoventral and anteroposterior, which will determine the regional cell identity within the neural tube. Serotonergic (5-HT) neurons appear around E10–E11.5 and are induced by morphogens like Fgf8 and Shh227,230–232. The 5-HT neurons will continue to developing through E12.5 233 . For 5-HT neurons to undergo conversion from 5-HT progenitors to 5-HT precursors, and then to postmitotic 5-HT neurons, several transcription factors are required. These transcription factors are induced by Fgf8 and Shh (Fig. 4C). Shh induces the expression of transcription factors including Nkx2.2, Nkx6.1, Foxa2, and Mash1 (Ascl1) in the 5-HT progenitor cells. As these transcription factors are expressed, they activate other specification factors like Pet1, Lmx1b, Gata2, Gata3, and Insm1 that are found within 5-HT precursor cells227,231. Lmx1b is likely positioned upstream of Pet1 and necessary for the maintenance of Pet1 but is not required for Pet1 activation. Pet1 is essential for the terminal differentiation of 5-HT precursors to convert to postmitotic 5-HT neurons that express aromatic L-amino acid decarboxylase (AADC) and TPH2 (Fig. 4C)227,234. Certain transcription factors such as Nkx2.2, Nkx6.1, and Gata2 are important for neurogenesis, while Lmx1b and Pet1 are important for differentiation of the DRNs 231 . An additional morphogen signaling pathway that drives serotonergic development is a Wnt signaling pathway, the planar cell polarity pathway, which is specifically involved in axon development and directed cell migration235,236. Specifically, Wnt5a and Wnt7b are expressed in the brainstem and lead to directional cues with Wnt5a attracting serotonergic axons (Fig. 4C) 235 . The Wnt and planar cell polarity pathway aid in serotonergic organization throughout development. When the rostral serotonergic neurons undergo induction by morphogens, neurogenesis and differentiation pathways lead to the development of either the rostral cluster with rhombomeres 1–3 or caudal cluster with rhombomeres 5–8.
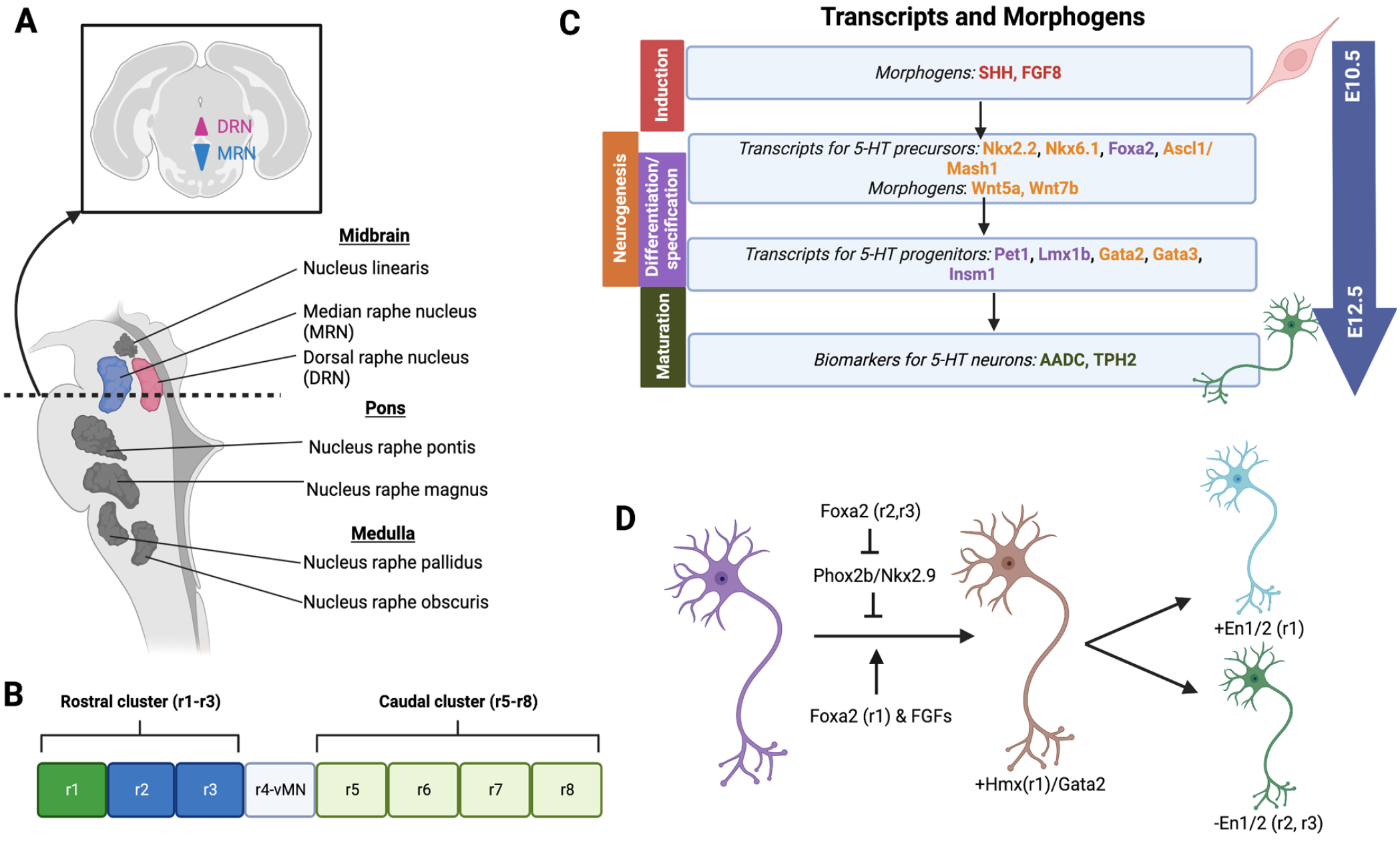
Development of serotonergic neurons with their associated transcription factors and morphogens. (A) The raphe nuclei are located in the midbrain with the MRN in blue and the DRN in pink. (B) There are eight rhombomeres that compose the rostral and caudal clusters. The MRN and DRN develop from the rostral cluster (r1–r3). (C) Within the rostral cluster there are various transcription factors and morphogens that contribute to the induction (red), neurogenesis (orange), differentiation/specification (purple), and maturation (green) of the serotonergic neurons as they develop from serotonergic (5-HT) progenitors to 5-HT adult neurons. The large blue arrow to the right indicates the embryonic days, E10.5–E12.5, that these neurons develop. (D) The interaction of several transcription factors involved in rostral cluster development of 5-HT neurons. The development relies on both inhibition and induction of transcripts and genes. MRN: median raphe nucleus; DRN: dorsal raphe nucleus.
The scope of this review will cover the developmental neurobiology of the rhombomeres 1–3 that give rise to the adult DRNs or median raphe nuclei, as these are the 5-HT neuron regions often used for transplantation studies and may provide a valuable source of 5-HT neurons for AD therapy. The presence of Foxa2 and repression of ventral motor neuron activators like Phox2b and Nkx2.9 likely contribute to the intrinsic determination of 5-HT neurons, as this pathway is activated in both the rostral and caudal rhombomeres. The rostral rhombomeres appear 1 day earlier in development, allowing them to proceed down a different cell fate pathway. The expression of Hmx homeodomains, Foxa2, and Engrailed (En1/2) genes distinguished rhombomere 1 from rhombomere 2/3 (Fig. 4D) 227 . More recent studies have been exploring additional signaling pathways and using single-cell RNA sequencing (scRNA seq) to unpack the developmental neurobiology of 5-HT neurons. The reelin-dab1 pathway mediates the lateral migration of the lateral wings of the DRNs. When reelin signaling was disrupted, lateral migration of the lateral wings was arrested. In addition, the serotonergic projections to brain structures such as the hippocampus, neocortex, and cerebellum were impaired, meaning reduced density of projections in superficial and deep plexuses 237 . scRNA seq analyses have identified a vast number of neuropeptides, serotonin receptors, neuropeptide receptors, and transcription factors that are distinct to the serotonergic neuron clusters. Notably, the co-expression of genes in functional gene categories has illuminated the connections between 5-HT neuron-associated genes, and whether they are involved in transcriptional regulation, synaptic connectivity, or neuronal communications 238 . These genes provide more information on the developmental neurobiology of the DRNs and median raphe nuclei, which may give insight into the novel molecular targets for the generation of exogenic 5-HT neurons through methods such as blastocyst complementation.
Developmental Neurobiology of Limbic Interneurons
A significant population of GABAergic interneurons arise from progenitor cells in the ganglionic eminences, which then migrate throughout the brain. After migration, interneurons form some of the first functional synapses in the brain and influence the development of neural circuitry 239 . Interneurons are an extremely heterogeneous class of neurons and their development, especially the determination of specific interneuron subtypes, is complex. For example, varied transcription factor expression denotes at least 18 interneuron progenitor domains in the subpallial ventricular zone 240 . Approximately 60% of interneuron progenitors originate in the MGE, express Nkx2.1 and Lhx6, and preferentially develop into fast-spiking PV+ and non–fast-spiking SST+ cells. Sox2, Otx2, and potentially Hhex are also required for appropriate MGE formation196,241,242. Similarly, Emx1/Emx2 play a role in broad forebrain specification and can be successfully targeted to generate some populations of donor-derived interneurons in the forebrain via blastocyst complementation23,243. Approximately 30% of interneurons originate in the caudal ganglionic eminence (CGE) and are biased toward VIP+, calretinin+, calbindin+, and Reelin+ cell fate. The gene Foxg1, also known as Bf1, is necessary for the development of both MGE- and CGE-derived interneurons 244 . The remaining 10% of interneurons include progenitors from the POA, which form a variety of interneuron subtypes, and other, less studied cells from the lateral ganglionic eminence (LGE)154,155,240,245–247.
Nkx2.1 is a key gene for interneuron development with expression beginning around E9.5 in regions overlapping with Olig2, and different interneuron subtypes emerge from Nkx2.1+Olig2+ progenitors in a temporal fashion (Fig. 5) 248 . Foxg1-driven Olig2 ablation does not affect the differentiation of interneuron subtypes 248 . However, maintenance of Nkx2.1 expression between E10.5 and E12.5 appears crucial for PV and SST interneuron specification. A conditional knockout of Nkx2.1 between E10.5 and E12.5 resulted in nearly total loss of PV+ and SST+ interneurons, some of which were re-specified to CGE-derived subtypes155,249. Nkx2.1-deficient mice have an approximate 40% reduction in GAD67+ cells at E18.5 250 . Thus, Nkx2.1 is essential for appropriate induction of interneuron progenitor cells and may be a potential target to produce exogenic interneurons, especially PV+ and SST+ interneuron subtypes.
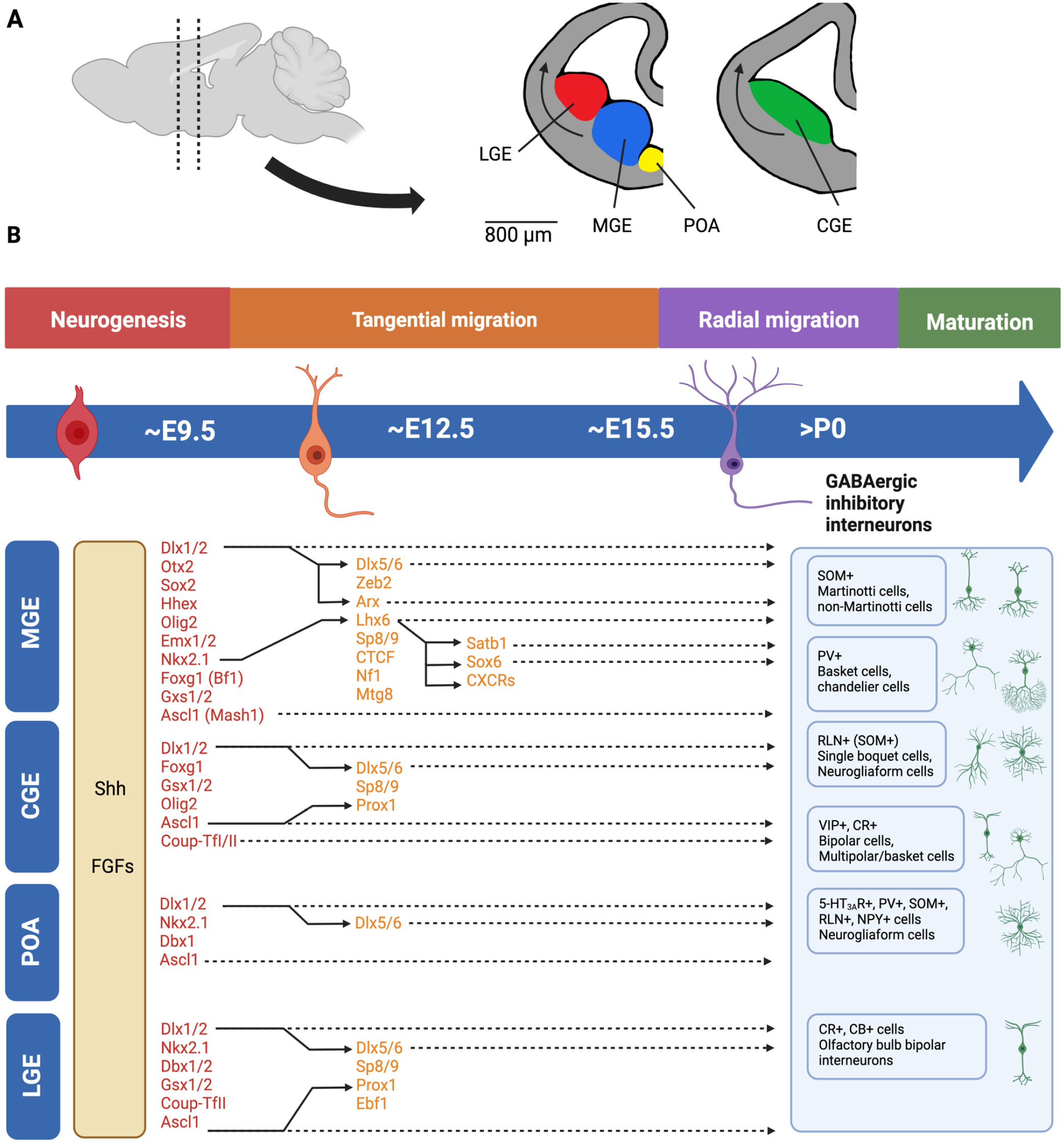
Development of interneurons. (A) Interneurons originate from subpallial progenitor domains including the MGE, CGE, LGE, and POA. Beginning around E12 and E13, maturing interneurons migrate tangentially through the brain to the cortex and limbic system. (B) Approximate timeline of the induction (red), tangential migration (orange), radial migration (purple), and maturation (green), including the differentiation/subtype specification of interneurons, controlled by a variety of transcription factors (colored text to match approximate timeline of development) and morphogens (orange-brown box). MGE: medial ganglionic eminence; CGE: caudal ganglionic eminence; LGE: lateral ganglionic eminence; POA: preoptic area.
Genes downstream of Nkx2.1 may also be potential targets, particularly Lhx6. Nkx2.1 is known to induce Lhx6 and Lhx8 activity. Lhx6 specifies GABAergic interneuron, while Lhx8 determines cholinergic fate154,155,251. A targeted disruption of Nkx2.1 led to a lack of Lhx6 and Lhx8 expression at E12.5 250 . Nkx2.1 and Lhx6 have been considered “master regulators” of interneuron development154,252. Lhx6-expressing cells are first observed at E11.5, and are strongly expressed in the MGE between E12.5 and E16.5 with continued expression into adulthood252–254. This gene is required for migration, particularly for PV and SST cells252–254. Ablation of Lhx6 results in abnormal interneuron migration and distribution in the cortex, and MGE cells undergo apoptosis or acquire a CGE-like fate255–257. The transcriptional repressor CTCF is important for maintaining Lhx6 expression in maturing interneurons. A conditional ablation of CTCF in MGE cells led to reduced Lhx6 expression, delayed migration, and a reduction of PV+ and SST+ cells with an apparent fate switch to Lhx8 projection neurons252,257. Lhx6 expression is also regulated by the transcription factor Sp9, which is expressed in the MGE at E13.5 258 . Ablation of Sp9 results in a roughly 50% loss of MGE-derived cortical interneurons 258 . Sp9 and the related factor Sp8 were also implicated with development of calretinin+, reelin+, and VIP+ CGE-derived interneurons and calretinin+ and calbindin+ LGE-derived interneurons of the olfactory bulb259–261. More recent studies showed that Lhx6 interacts with Mtg8, which affects development of primarily SST+ and NPY+ interneurons, and Nf1, which affects PV+ interneurons262,263.
Other genes are also upregulated as interneuron progenitors migrate from the subpallium throughout the brain. These include Gsx1/2, historically also known as Gsh1/2. Gsx1/2 are important for MGE-, CGE-, and LGE-derived interneuron development264,266. Gsx1/2 are known to drive Mash1 (Ascl1) expression, and also interact with Dlx1/2, which can inhibit Nkx2.1 and activate Dlx5/6 to specify interneuron fate94,155,267,268. Mash1 can induce Prox1, which is expressed across the MGE, CGE, and LGE at E10.5. Prox1 is enriched in the CGE at E14.5 and is associated with CGE determination and migration155,264,269. Mash1 also has an early role in interneuron development by regulating early ventral telencephalon neurogenesis. Mash1−/− mice have defects in the MGE and LGE, reduced Nkx2.1 expression at E12.5, and disrupted timing of Gad67 and Dlx gene expression155,270. Dlx1/2 are first expressed between E9.5 and E10.5 and maintained into adulthood. These genes are especially important for interneuron progenitor migration and survival271–273. No detectable migration of GABAergic progenitors is seen in Dlx1/2 mutants at E12.5 or E15, and there is a significant lack of GABA+, calbindin+, and GAD67+ cells in the cortex at postnatal day 0 274 . Dlx1/2−/− double-mutant mice demonstrate significant deficits in the LGE and partial deficits in the septum, while Dlx1/2−/−: Ascl1−/− triple mutants had broad deficits in LGE and septum 267 . Dlx family transcription factors are also involved in interneuron subtype specification. PV+ interneurons express Dlx5/6 but not Dlx1, and loss of Dlx5 alone or in combination with Dlx6 led to PV+ interneuron deficits155,271,275. A population of Lhx6−Dlx+ migrating interneurons appear to be CGE and/or LGE derived. Another transcription factor from the Dlx family, Dlx2, can regulate calbindin+ cell development via the protein Necdin240,276,277. The gene Zeb2, also known as Sip1 or Zfhx1b, acts downstream of Dlx1/2 and is important for determining MGE-derived interneuron fate and migration278,279. The genes Coup-TfI/II regulate the balance between MGE and CGE fate. Coup-TfI/II are implicated in migration and specification of CGE interneurons, which was determined by a Dlx5/6-driven Coup-TfI knockout that had decreased CR+ and VIP+ cells, with a corresponding increase in PV+ cells280,281.
The gene Arx also appears to be important for later interneuron development. Expression begins around E10.5, continues to be expressed in a majority of migrating interneurons at E15.5, and is maintained in a subpopulation of mature interneurons after birth 245 . Disruption of Arx leads to defects in the basal ganglia and abnormal migration of interneurons, and one study observed loss of cortical and hippocampal interneurons at E18.5282,283. Lhx6 and Dlx2 can drive Arx expression by binding upstream enhancers, thus by supplementing Lhx6−/− animals with Arx normal phenotypes can partially be restored256,284. Similarly, both Lhx6 and the Dlx genes are upstream of chemokine receptors CXCR7 and CXCR4, which are important for interneuron migration256,285. Additional genes downstream of Lhx6 that affect late embryonic interneuron development, especially of PV+ and STT+ cells, include Sox6 and Satb252,286,287. In Sox6 null animals, development of PV+, SST+, basket cells, and Martinotti cells is abnormal 288 . Based on these previous genetic ablation studies, genes expressed later in interneuron development may be potential targets for generating exogenic interneurons specific to a particular subtype or location in the brain.
Development of interneurons is also influenced by morphogens like Shh and FGFs. As with cholinergic neurons, Shh appears to act upstream of Nkx2.1 to help define the MGE. Specifically for interneurons, Nkx2.1 also acts upstream of Dlx2 and Gsx2 289 . Interestingly, Nkx2.1-deficient mice have reduced Shh starting at E8.75 and at E11.5, and Lhx6 can promote Shh, suggesting a feed-forward mechanism of specification250,252,290. Shh is also involved in the specification of the other interneuron progenitors. Shh prevents upregulation of Gsx2, promoting a CGE-like, calretinin+ fate, and also promotes SST+ interneuron fate at expense of PV+ interneurons in the MGE291,292. FGF receptors 1–3 are expressed in ventral telencephalon progenitors, and Fgfr1 alone appears to be the most sufficient for ventral patterning. Fgfr1- and Fgfr3-mutant mice lack Ebf1, typically expressed in the LGE, and later have reduced NPY expression. Fgfr1 and Fgfr2 mutants lack Shh, Ebf1, and MGE-associated transcription factors Lhx6 and Lhx8, and later have reduced Gad67 expression 287 . These results suggest that Shh and the FGFs are involved in complex pathways, which promote both the induction and differentiation of interneuron progenitors. Therefore, a number of important transcription factors and morphogen pathways including but not limited to Nkx2.1, Lhx6, Dlx1/2, Arx, and Shh should be considered candidate genes for generating exogenic interneurons using blastocyst complementation.
Target Genes for Knockout to Create Developmental Niches for Exogenic Neurons
Candidate genes for specific organs, tissues, and cells may provide developmental niches for the targeted organ, tissue, or cell of interest to be generated through blastocyst complementation. The aforementioned neuronal cells provide reasonable exogenic sources for alleviating AD neuropathology, and thus AD-associated cognitive impairments. Based on the neurodegeneration, the success of transplantation, and developmental neurobiology for exogenic neurons of interest, candidate genes have been compiled as targets for genetically engineering a developmental niche for the gene’s associated cell type (Table 2). Depending on the region and or cell type of interest, one or more target genes may be viable candidates. In certain cases, a niche may be better specified by a combination of knockouts.
Gene KO Targets for Generation of Exogenic Neurons.

BMP: bone morphogenetic protein; FGF: fibroblast growth factor; Shh: sonic hedgehog.
Conclusion
The progressive neurodegeneration and dementia that accompany AD are devastating. Current AD therapies are rather limited, often only alleviating symptoms and not stopping the course of the disease. Thus, novel technologies and therapies for AD must be developed. Transplantation of healthy exogenic neurons to supplement and restore neural activity may provide a novel approach for AD therapy. Blastocyst complementation offers a promising approach to generate a renewable source of neurons that develop with an in vivo context and recapitulate the neuron-specific characteristics and physiology. This versatile technology can be adapted to generate a variety of neural cells affected by AD, including hippocampal neurons and limbic projection neurons, cholinergic nucleus basis and medial septal neurons, noradrenergic LC neurons, serotonergic raphe neurons, and limbic interneurons. Consideration of the developmental biology of each of these neuronal cell types is critical for determining appropriate candidate genes for blastocyst complementation. While the blastocyst complementation approach is promising, the formation of interspecies chimeras still has its challenges. Interspecies chimeric barriers such as asynchronous developmental timing and cell–cell competition limit the efficiency and competency of generating exogenic cells and tissues14,293. Ongoing and future studies aim to investigate the molecular mechanisms behind these barriers and provide insight into strategies to improve chimeric competency294–296. With a better understanding of interspecies chimeric barriers and developmental pathways, blastocyst complementation opens a novel approach and therapy method for treating neurodegenerative diseases like AD.
Footnotes
Author Contributions
All authors confirm contribution to the preparation and revision of this manuscript, and approved the final version of the manuscript.
Availability of data and material
Not applicable.
Ethical Approval
This study was approved by our institutional review board.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by grants from Regenerative Medicine Minnesota, the National Institutes of Health (R01 AI173804), and from the Suzanne M. Schwarz Fund.