
Abstract
Blastocyst complementation combined with gene editing is an emerging approach in the field of regenerative medicine that could potentially solve the worldwide problem of organ shortages for transplantation. In theory, blastocyst complementation can generate fully functional human organs or tissues, grown within genetically engineered livestock animals. Targeted deletion of a specific gene(s) using gene editing to cause deficiencies in organ development can open a niche for human stem cells to occupy, thus generating human tissues. Within this review, we will focus on the pancreas, liver, heart, kidney, lung, and skeletal muscle, as well as cells of the immune and nervous systems. Within each of these organ systems, we identify and discuss (i) the common causes of organ failure; (ii) the current state of regenerative therapies; and (iii) the candidate genes to knockout and enable specific exogenous organ development via the use of blastocyst complementation. We also highlight some of the current barriers limiting the success of blastocyst complementation.
Introduction
In the 2017 year-end annual report, the United States Department of Health and Human Services Organ Procurement and Transplantation Network reported that nearly 115,759 patients needed lifesaving organs, but only 34,770 transplants were performed. For many end-stage diseases, organ transplantation is the final resort for survival after all other treatments have been rendered ineffective. The severe shortage in the availability of organs and the increase in their demand have forced the research community to develop novel sources of organs and cells for transplantation in humans. Various in vitro techniques have been described for the differentiation and maturation of numerous cell types in both two-dimensional cultures and three-dimensional “organoid” cultures. While many of these protocols have resulted in cell lines that can mimic various properties of their in vivo counterpart, they cannot yet recapitulate the full spectrum of spatiotemporal signals required to develop fully functional tissues and organs for transplantation.
One approach that may have immense potential in addressing the shortage of organs for transplantation is ‘blastocyst complementation’ (Fig. 1). In this approach, embryos from one organism are genetically engineered so that they lack a functional gene(s) necessary for the development of the tissue of interest. The organogenesis-disabled embryos are then microinjected with healthy pluripotent stem cells (PSCs) from a second organism, and are then transferred into a maternal surrogate. Through normal mammalian development, the microinjected PSCs occupy the niche left by the gene knockout, and the progeny of these cells develop into a functional organ. Beginning in the early 1990s, with a resurgence in the last decade, blastocyst complementation has surpassed many milestones with the goal of generating human organs within non-human hosts (Table 1). Currently, the pig is the model of choice for chimeric host organism due to anatomical and physiological similarities with humans 7 –9 . Gene editing technologies such as zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the clustered regularly interspaced short palindromic repeats/Cas9 (CRISPR/Cas9) can be used to inactivate (knockout) specific genes in mammalian cells at high efficiency 10 . Specifically, in the pig, genomic editing was achieved using both TALEN 11 and CRISPR/Cas9 12 approaches.

Cartoon schematic of blastocyst complementation. Human pluripotent stem cells grown in vitro are microinjected into genetically engineered porcine blastocysts which are then transferred to surrogate sows. The chimeric blastocysts will develop to a fetal stage in which human neuronal stem/progenitor cells can be harvested or to live-born animals where mature human organs can be harvested and processed for transplantation into patients.
Milestones of Blastocyst Complementation.

It is highly conceivable that blastocyst complementation can be used for the generation of human pancreas, liver, heart, kidney, lung, skeletal muscle, as well as cells of the immune and nervous systems. Within this review, we will focus on the development of these tissue systems and identify candidate genes for use in blastocyst complementation (Table 2). Knockout of candidate genes should, in theory, create a vacant niche in the organ/tissue system of interest for cells derived from the microinjected PSCs to occupy and proliferate, leading to fully functional tissues. Furthermore, candidate genes should be spatially restricted to limit the potential for off-target chimerism.
Candidate Genes for Blastocyst Complementation.
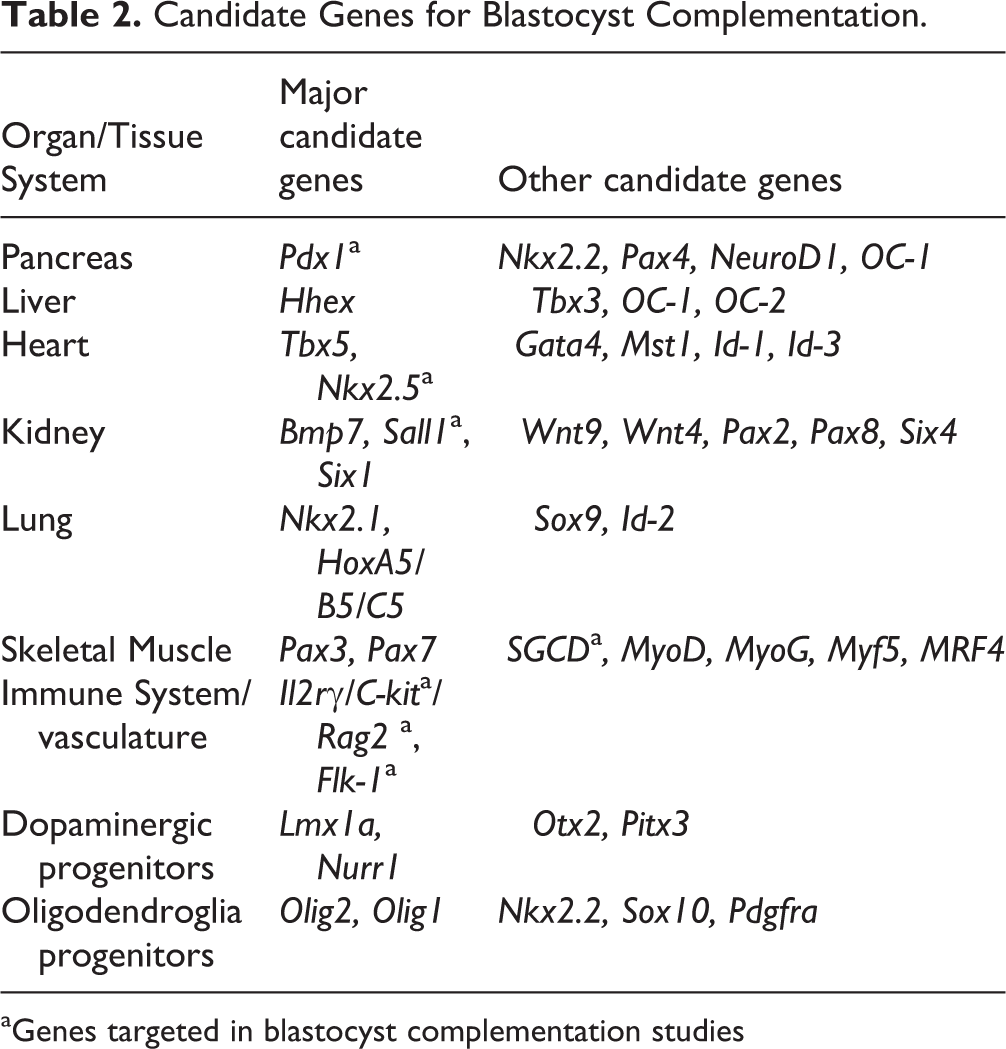
aGenes targeted in blastocyst complementation studies
Pancreas
The human pancreas performs essential exocrine and endocrine functions. Pancreatic acinar cells produce enzymes critical for food digestion, and hormones from the endocrine cells of the pancreatic islets control blood glucose homeostasis. Medical conditions, such as diabetes and pancreatitis, that result in pancreatic dysfunction are extremely debilitating for patients and place an increasingly untenable economic burden on healthcare services and society. However, current treatment options for pancreatic diseases are limited. Currently, the pancreatic disorder that urgently requires new treatment options is type 1 diabetes mellitus (T1DM). T1DM is an autoimmune disorder that results in the destruction of the insulin-secreting β-cells affecting more than 30 million Americans 13 . Despite improvements in life-sustaining exogenous insulin therapies currently available, such as insulin pumps and continuous glucose monitoring systems, treatment via this route is non-physiologic resulting in secondary complications 14,15 .
Allogeneic human pancreatic islet transplantation has demonstrated value as a therapeutic modality for T1DM that can restore insulin independence by providing a physiologic insulin source resulting in near-normoglycemia 16,17 . Due to the limited supply of suitable donor organs, however, alternative sources of pancreases or islets are needed to extend these therapies to the vast majority of patients with T1DM. Although there has been great interest in the development of virtually unlimited sources of islets, such as porcine islets or β-cells derived from human pluripotent stem cells, each of these methods still has major technical hurdles to overcome before they are ready for widespread clinical use 18 .
Alternatively, blastocyst complementation may provide a route to a virtually unlimited supply of human pancreas and make pancreatic islet transplantation widely available to patients with T1DM. In fact, blastocyst complementation has successfully generated a functional pancreas in mice, rats, and pigs through blastocyst complementation targeting of the Pdx1 gene 2 –5,19 . Functional PDX1 activity is critical for the development of the pancreas, beta cell maturation, homeostasis between neogenesis and apoptosis, duodenal differentiation, and biliary development. Hence, Pdx1 represents an obvious target for gene knockout and blastocyst complementation 20 . Through TALEN or CRISPR/Cas9 genome editing, Pdx1 mutations have previously been introduced into donor mouse or rat embryos 4,5 . Similarly, porcine fibroblasts genetically engineered to express the transcription factor HES1 from the PDX1 promoter repressed pancreatic development in pigs following somatic cell nuclear transfer 3 . The pancreatic disabled embryos from these studies were injected with induced pluripotent stem cells (iPSCs) from the same species (mouse → mouse, rat → rat, pig → pig) or from different species (rat → mouse, mouse → rat) 2 –5 . The resulting chimeric offspring presented with pancreata comprised of donor cells, which were sensitive and responsive to glucose. Furthermore, glucose tolerance was restored in diabetic mice following transplantation of islet cells derived from mouse → rat chimeras 4 .
Other transcription factor candidates involved in fetal development of the pancreas may provide alternative host niches for either generation of the entire pancreas or more targeted pancreas tissue replacement using blastocyst complementation. Additional candidate genes for knockout include Nkx2.2, Pax4, Neurod1, and OC-1. Inactivation of genes involved in neogenesis such as Pdx1 and Nkx2.2 resulted in a substantial and complete loss of alpha and beta islet cells. Pax4, known to be associated with the maturation of beta cells, may also provide a holistic niche for human stem cells to propagate. Neurod1 inactivation results in reduced beta cell populations and the inability to produce mature islets. This gene has also shown involvement in both pancreatic cell expansion and glucagon expression. In terms of addressing the exocrine component of the pancreas, OC-1 is expressed in early and late pancreatogenesis and is significantly involved in the development of the pancreatic duct 21 .
Liver
The liver is the largest organ in the human body, and has the unique potential to regenerate its entire mass even after major resection 22,23 . It also performs a wide variety of complex and critical metabolic, synthetic, immunologic, and detoxification functions. In 2016, more than 40,000 individuals died as a result of chronic liver disease and cirrhosis 24 . Alcohol consumption, obesity, type 2 diabetes, metabolic syndrome, and infection with hepatitis viruses are major causes of acute and chronic liver diseases, as are many inborn errors of metabolism 25 . Even though hepatocyte transplantation could potentially correct a variety of these disorders, whole liver transplantation remains the most viable and appealing option.
Due to the severe shortage of livers for transplantation, efforts are being made to restore the function of damaged livers by alternative means, including hepatocyte transplantation 26,27 . A number of techniques have been developed to optimize the growth and propagation of primary and stem cell-derived hepatocytes in vitro, and to maintain their phenotypic characteristics 28 –30 . Investigations aimed at culturing hepatocytes as 3D spheroids have demonstrated improved function when compared with monolayer cultures 31,32 , which can be further augmented using arrays 33 , bioreactors 34 , microencapsulation 35 –37 , and decellularization/recellularization of biologic scaffolds 38,39 . Successful hepatocyte transplantation, however, requires reliable sources of hepatic cells (both intrahepatic and extrahepatic). High-level propagation of primary hepatocytes that ensures viability and maintenance of phenotypic characteristics of in vitro expanded hepatocytes, as well as efficient hepatic differentiation protocols, are major roadblocks to achieving clinical success.
The challenges to generating large quantities of hepatocytes can be directly addressed, in part, by creating human-animal liver chimeras via blastocyst complementation. In the mouse embryo, the liver is first detected as an outgrowth bud of proliferating endodermal cells in the ventral foregut on day 8 of gestation 40 . Beyond the induction stage, various transcription factors are needed for endoderm patterning and organ development. Among these, the hematopoietic-specific expression of HHEX plays a pivotal role in liver development 41 , where it functions both as a transcriptional repressor and activator 42,43 . It is highly conserved in human, mouse, Xenopus, zebrafish and other multicellular organisms 44 . Disruption of the Hhex gene has been shown to result in embryonic lethality in mice 41,45 –47 . Analysis of Hhex-knockout mice demonstrated the initial formation of the liver bud, which is then arrested beyond E9.5, suggesting that Hhex is an ideal candidate for blastocyst complementation 41,45,48 .
Studies using mutant mice have also shown that hepatoblast differentiation into biliary epithelial cells, which develop into the biliary tree, can be prevented through Tbx3 inhibition of notch signaling 49,50 . Another set of transcription factors, OC-1 and OC-2, promote the expression of α2-macroglobulin and follistatin, inhibitors of the TGF-β/activin pathway that is critical for the formation of the biliary tree 51 . During liver development, OC-1 and OC-2 also promote the migration of hepatoblasts in the septum transversum by stimulating the degradation of the basal lamina surrounding the liver bud 52 . Unbalanced expression of these transcription factors could lead to impaired migration and enhanced cell clustering.
Heart
Diseases of the heart are the leading cause of death in the United States, responsible for over 600,000 in 2015 53 . Dilated cardiomyopathy (DCM) and coronary artery disease (CAD) are the primary indications for heart transplant, accounting together for over 94% of the heart transplants performed in the US in 2015 54 . Heart transplantation is the definitive cure for DCM and coronary artery bypass graft and percutaneous coronary intervention are revascularization procedures commonly used to treat CAD 55 . Significant advances in immunology, immunosuppression, as well as peri- and post-operative management have dramatically reduced morbidity and mortality related to heart transplantation. Despite these improvements, heart transplantation is still associated with several limitations. Primary among these limitations is the need for donor hearts, which has steadily increased since 2007 and has risen 51% overall since 2004 54 .
Blastocyst complementation could potentially be used to generate exogenous human hearts, but it is essential to understand the complex process of cardiogenesis, which involves several key genes, including Tbx5, Nkk2.5, Gata4, and Mst1. The T-box-containing transcription factor TBX5 has a critical role in cardiogenesis and septation. Overexpression of a dominant-negative form of the Tbx5 gene in mice results in a severely abnormal or absent heart 56 . Both TBX5 and NKX2.5 interact to promote differentiation of cardiomyocytes 57 . GATA4 is a zinc finger transcription factor and is one of the earliest transcription factors expressed by developing cardiac cells. While Gata4 is not essential for specifying a cardiac lineage, it is a key regulator of embryo folding. Gata4-null mice have generalized severe defects in ventral body patterning that leads to a failure to form a linear heart tube resulting in embryonic lethality between E7.0 and E10.5 58,59 . Mouse studies have demonstrated GATA4 physically interacts with NKX2.5 to synergistically increase atrial natriuretic factor expression 60 . GATA4 also interacts with TBX5, and mutations in Gata4 can disrupt this binding, causing congenital heart defects in humans 61,62 .
Overexpression of pro-apoptotic factor MST1 in mice results in cardiac-specific apoptosis and consequently adult-onset cardiomyopathy in mice. Complemented Mst1 mutant blastocysts with healthy embryonic stem cells (ESCs) resulted in chimeric mosaicism of the heart with cells derived from both host and donor. The hearts of the chimeric mice functioned normally, with cell densities consistent with those of wild-type mice 63 . Other key components of cardiogenesis are the Inhibitor of DNA binding proteins 1 and 3 (ID1 and ID3) that are expressed during mid-gestation and are responsible for the formation of the epicardium, endocardium, and cardiac valves. Ablation of Id1 and/or Id3 results in cardiac defects and lethality in mice. Injection of healthy murine ESCs into Id1 and Id3 knockout blastocysts was able to rescue heart functioning to normal functioning levels and correct for otherwise altered gene expression profiles 64 .
Finally, the homeobox-containing gene Nkx2.5 plays an essential role in cardiac development and may be a viable target for blastocyst complementation 65,66 . It is expressed early in heart development and regulates the expression of multiple downstream genes involved in cardiogenesis 67 . Mice with homozygous Nkx2.5 mutations have abnormal cardiac looping, stunted growth, and die between E9.5 and E11.5 67,68 . Complementing Nkx2.5 mutant mouse blastocysts with rat iPSCs has been shown to rescue cardiac development and embryo growth at E10.5, but it has not yet been demonstrated to produce a live rescued chimera 5 . Given the many interactions between the genes involved during cardiogenesis, it may be necessary to identify multiple genes for successful organ generation via the approach of blastocyst complementation.
Kidney
According to the National Kidney Foundation, 30 million adults in the United States have chronic kidney disease (CKD) 69 . CKD arises from abnormalities in kidney structure and function that produce a gradual decline in glomerular filtration rate over months to years. In the United States, diabetes mellitus and hypertension are the leading causes of CKD 69 . Other etiologies include polycystic kidney disease, congenital abnormalities of the urinary tract, autoimmune diseases like systemic lupus erythematosus, and primary glomerulonephritis such as IgA nephropathy 70 . Patients with CKD have reduced life expectancy and increased risk of cardiovascular disease 70 . Progression of CKD may result in end-stage kidney failure requiring renal replacement therapy. Dialysis is life-saving but does not produce the same longevity as people with normal functioning kidneys 71 . Besides the high financial burden associated with chronic dialysis, the 5-year survival rate of patients on dialysis is only 36% 72 . Kidney transplantation offers the most effective treatment to restore kidney function but is limited by a severe organ shortage. One potential solution is to generate kidneys via the approach of blastocyst complementation.
Kidney development is a complex process involving reciprocal signaling between the branching ureteric bud, which gives rise to the collecting system, and the adjacent metanephric mesenchyme, which gives rise to the nephrons. Many growth factors, receptors, and transcription factors that are essential for normal kidney development have been identified. Thus, there are many candidate genes for kidney organogenesis via blastocyst complementation. For example, bone morphogenic protein-7 (BMP7) is a potential candidate gene because of its essential role during embryogenesis. BMP7 is expressed in the metanephric mesenchyme and ureteric buds 73 , and knockout mice that are deficient in BMP7 show severe kidney abnormalities. Absence of BMP7 affects the expression of molecular markers of nephrogenesis, such as PAX2 and WNT4, between embryonic days 12.5 and 14.5 74 . Both PAX2 and PAX8 are required for branching morphogenesis and nephron formation 75 . WNT genes are critically important in embryonic kidney development as they control ureteric bud formation, nephrogenesis, and stem cell renewal. WNT9-deficient mice die within 24 h of birth due to kidney agenesis, and WNT4-deficient mice die due to a failure of epithelial development 76 . Inactivation of SIX1 in porcine fetuses resulted in failure in the development of the branching ureteric bud, and dual inactivation SIX1/SIX4 arrested kidney development at metanephros formation 77 .
Another essential gene that may be a candidate for blastocyst complementation is Sall1. SALL1-deficient mice die during the perinatal period from kidney agenesis 78 . The expression of SALL1 in the metanephric mesenchyme surrounding the ureteric bud further suggested that it plays an essential role in metanephros development. Blastocyst complementation has been successfully used to generate PSC-derived mouse kidneys in Sall1 mutant mice 78 . In this procedure, both mouse ESCs and mouse iPSCs were capable of forming a kidney within Sall1-deficient mice. Flow cytometry and PCR confirmed the cell lineages, and histological analysis indicated that the injected PSCs in SALL1-deficient chimeric mice replaced the kidney epithelial cell lineages of the metanephros. Similarly, complementation of Sall1-deficient rats with mouse ESCs resulted in kidneys comprised entirely of mouse cells expressing markers indicative of functional tissue 79 .
Lung
Lung disease is a major cause of pulmonary morbidity and mortality. When considering the major lung diseases where lung transplantation is a viable option, 81% are either chronic obstructive pulmonary disease (COPD), interstitial lung disease (ILD) with or without pulmonary fibrosis, and bronchiectasis with or without cystic fibrosis 80 . The most common disease of the lung, COPD, is characterized by a general restriction of airflow to the lung tissue and leads to frequent coughing, sputum production, and shortness of breath 81 . Conversely, ILD represents many different diseases and complications, most of which are related primarily to malformations and fibrosis of the connective tissue surrounding alveoli within the lung. Restriction of air passage leads to mild to extreme shortness of breath and a painful dry cough 82 . Excessive fibrosis and scarring can then lead to pulmonary fibrosis, a more serious condition where lung transplantation becomes necessary 83 .
Between 1990 and 2013, there were 45,452 lung transplantations reported globally to treat progressive lung disease. This large cohort had a median survival of 5.7 years after transplant and a survival rate of 31% at 10 years post-transplant. The need for an alternative source of lung tissue is clear, as there were 4218 lung transplants reported to the International Society for Lung and Heart Transplantation in 2015, with many more left on donor waiting lists 84 .
Generation of lung tissue has not yet been attempted by blastocyst complementation, in part because there is no clear gene candidate for inactivation. For this reason, development of a lung may require a combination of different gene and transcription factor knockdowns. Currently, the candidate gene with the most potential as a regulator of overall lung development knock-down is Nkx2.1, given that it is the first recognizable transcription factor present during the onset of the early lung buds and Nkx2.1-null mice lack lung formation 85 . At E9.0 in mice, and 28 days gestation in humans, NKX2.1 is expressed along with LEF1 and AXIN2 on the ventral side of the anterior foregut endoderm and is driven upstream by WNT/β-catenin signaling 86 . Shortly after initial NKX2.1 detection, expression increases and a primitive trachea extends followed by formation of the two lung buds into the mesenchyme. The proximal-distal axis is initially regulated by SOX2 expression at the proximal end and SOX9/ID2 expression at the distal end and is responsible for the migrating split at E12.5 in the mouse. The proximal SOX2 progenitor cells differentiate into secretory and ciliated lineage cells 87 , whereas the distal SOX9/ID2 progenitor cells differentiate into type 1&2 terminal alveolar cells 88 . HOX5 is an external regulator of WNT signaling in the patterning of the lung related to BMP4 feedback and NKX2.1. Triple paralog HOXA5/HOXB5/HOXC5 mutant embryos lose WNT2/2b signaling completely in the mesenchyme and have severe defects in proximal-distal patterning and overall branching 89 . Clearly, similar to all complex organs of the body, there are many different genes that play major roles in the development of the lung. Currently, Nkx2.1 in combination with all three Hox5 paralogs represent the best candidate genes for lung organogenesis through blastocyst complementation, as these are the early genes in initial lung bud formation.
Skeletal Muscle
Muscular dystrophies are one of the more prevalent diseases that affects skeletal muscles. The most common form of this affliction is Duchenne muscular dystrophy (DMD), in which symptoms manifest in the form of muscle weakness and can lead to insufficient respiratory muscle function and cardiomyopathy 90 . The disorder itself is a recessive genetic condition associated with the dystrophin gene. Two-thirds of cases are inherited from the parent as an X-linked recessive trait, while the rest are the result of mutations within the gene itself 91 . Muscles that lack functional dystrophin are more prone to injury and have been found to undergo cycles of necrosis and regeneration 90 . There is currently no cure for the disease, but gene therapies have created treatments that are helping to eliminate symptoms and recover some degrees of function 92 .
Muscle satellite cells are stem cells that can proliferate to produce myoblasts and regenerate muscle fibers. Muscle satellite cells are quiescent in adults, but, following injury, they are activated to express MYOD and begin the production of myogenic precursors cells for the repair and regeneration of muscle fibers within the body 93 . The potential of satellite cells to aid in the regeneration of muscle systems has led to increased studies for the role of transplantation as a treatment modality. Myoblasts can be isolated from adult muscles and expanded ex vivo 94 . Through this process, the myoblasts maintain their ability to further differentiate into muscle fibers and form connections between tissues, which presents unique opportunities for transplantation therapies. Myoblast transplantation has been successfully performed in both mouse models and patients suffering from DMD 93 . Diseased muscles fuse with the newly introduced myoblasts, leading to improvements in their function as the new cells integrate into the system and differentiate into healthy fibers 94 . However, satellite cells freshly isolated from adult muscles are best suited for regeneration due to their ability to engraft within the native muscle cells and self-renew 93 .
Myogenic regulatory factors (MRFs) are key in myogenic specification and characterizing the expression of muscles cells. There are four MRFs that drive myogenesis within muscle stem cells: MYF5, MRF4, MYOD, and MYOG 95 . Knocking out single MRFs or combinations of MRFs can lead to a total loss of body muscles as reported in mouse models. Two of the most important genes for muscle development are Pax3 and Pax7 as upstream genes of MRFs. Knocking out the Pax3 and Pax7 genes completely halts myogenic differentiation 95 .
Through blastocyst complementation, it is possible to induce the growth of human skeletal muscles that are fully functioning and provide satellite cells that produce healthy myoblasts. While they can be used as various therapies for a number of disorders, their greatest potential may be in the treatment of DMD 93 . Blastocyst complementation has been the focus of research to generate healthy skeletal muscle within animal models of muscular dystrophy. Microinjection of healthy mouse ESCs into blastocysts derived from the dystrophin mutation mdx mouse model of DMD resulted in chimeric mice, presenting with up to 30% mosaicism in skeletal muscle, and significantly increased levels of muscular dystrophin 96 . Similarly, microinjection of healthy mouse ESCs into blastocysts derived from the sarcoglycan-δ (SGCD) knockout mouse model of Limb-girdle muscular dystrophy-2F resulted in chimeric mice that were observed with a normal sarcoglycan complex within the skeletal and cardiac muscles 97 . Mosaicism of levels lower than 60% in the SGCD complemented mice, however, were unable to ameliorate the pathology seen in SGCD knockout mice 97 . While neither of these studies identified complementation of myoblasts, the degree of chimerism within the skeletal muscle of the mdx and SGCD mutant mice may provide targets for future complementation studies.
Immune Cells/Vasculature
Diseases of the immune system include severe combined immunodeficiencies (SCIDs), which are genetic diseases usually present at birth with non-functional T-helper cells combined with the absence of T-lymphocyte and B-lymphocyte function. X-linked SCID accounts for about 19% of all SCIDs and is due to a mutation in the IL-2Rγ gene 98 . Adenosine deaminase deficiency (ADA) makes up 10% of all SCIDs and causes accumulation of toxic dATP, which inhibits dNTP synthesis and resulting in the death of T-lymphocytes 98 . RAG-SCID (Omenn syndrome, OS-SCID) is caused by mutations of the recombinase activating genes (RAG1 or RAG2) that encode for enzymes involved in the first stage of V(D)J recombination and is critical to the immunoglobulin heavy-chain joining of B- or T-cell receptors accounts for roughly 15% of all SCIDS 98 .
Enzyme replacement therapy and hematopoietic stem cell (HSC) transplantation are the most common therapies for SCIDs. Although the success rate may reach 90% in matched HSC transplantation and 60–80% in haploidentical parent transplants, graft-versus-host disease remains a major concern in the context of incomplete HLA matching between donor and host. Blastocyst complementation could provide a source of HLA matched HSCs for use in the treatment of SCIDs.
A variety of SCID mice have already been developed that have aided in the study of the human immune system and disease, and can help to identify potential niches for blastocyst complementation 99 . The transgenic IL-2Rγ-deficient mouse presents with almost undetectable levels of circulating lymphocytes and NK cells. The c-kit point mutations lead to pleiotropic developmental defects, especially hematopoietic defects 100,101 . Chen and colleagues complemented RAG-2-deficient blastocysts with mouse ESCs to generate somatic chimeric animals with normal mature B and T cells, all of which were derived from the injected ES cells 1 .
Various techniques have been utilized to generate immunological chimeras. One method, currently employed by the Jackson Laboratory, is injection of human CD34+ hematopoietic stem cells into myeloablated adult non-obese diabetic SCID mice 102 . Within 12 weeks, the human CD34+ chimeric mice present with multi-lineage human hematopoiesis 102 . A second technique to generate chimeras is to introduce hematopoietic stem cells in utero. This method was published by Sasaki and colleagues, where Cynomolgus macaque ESCs were differentiated, in vitro, into a mesodermal lineage and injected into the primordial liver of sheep during the first trimester of development 103 . Low levels of macaque hematopoietic stem cells were observed in the peripheral blood and bone marrow up to 17 months following engraftment. While these two methods resulted in complementation of hematopoietic cells, a full recapitulation of the immune system was not observed.
Blastocyst complementation may be applicable to generate the full complement of immune cells. To test this hypothesis, Jansson and colleagues injected GFP-labeled mouse ESCs into W(41)/W(41) c-kit mutant mouse blastocysts, which typically present with reduced number and functionality of HSCs 104 . Fetal livers from the chimeric mice were isolated at embryonic day 14.5 chimeric mice, and GFP+ hematopoietic cells were observed at levels greater than 90%. Importantly, the dissociated cells from the chimeric GFP+ fetal liver were then transplanted into lethally irradiated mice, which resulted in near-complete engraftment in the bone marrow and peripheral blood. These results demonstrate an available niche for HSCs that can then be used therapeutically via blastocyst complementation.
A major obstacle in organ generation via blastocyst complementation is that the vasculature of the organ of interest is derived from cells from the host animal. In an effort to make a rejection-free transplantable organ through blastocyst complementation, it is necessary for the vasculature and the organ to be derived from human cells. The Nakauchi lab at the University of Tokyo has attempted to address this issue through complementation of Flk-1 mutant embryos 6 . Flk-1 is required for the formation of vascular and hematopoietic tissues. It was observed that complementation with mouse PSCs resulted in adult mice with donor-derived blood vessels and hematopoietic stem cells. While complementation of Flk-1 mutant embryos with rat PSCs did not result in live-born chimeras, these results provide an avenue for future complementation experiments where the risk of graft rejection is minimized.
Dopaminergic Neuron Progenitors
Transplantation of a population of dopaminergic neural progenitor cells as a therapy for Parkinson’s Disease (PD) has arguably advanced the furthest of any neurological cell replacement therapy, to date. The clinical diagnosis of PD occurs well into disease progression as patients will report symptoms of essential tremor, bradykinesia, and rigidity, which is a result of the loss of dopaminergic neurons within the substantia nigra 105 . While the prevalence of PD can be stratified among different geographical locations/ethnicities, it is generally estimated that almost 2 individuals per every 1000 in the population has symptoms which can be diagnosed as PD, with increased risk associated with aging 106 .
The success of cellular transplantation for PD is due to the identification of a population of dopaminergic neural progenitors within the fetal ventral mesencephalon (fVM), which can innervate the striatum and release dopamine to near physiological levels. Clinically, transplantation of fVM was attempted in the 1980s and 1990s in Europe and the USA. Patients were observed to benefit from transplantation with a reduction in off-medication Unified Parkinson’s Disease Rating Scale, an increase in radiolabeled 18F-DOPA uptake, and a reduction in anti-PD medication up to 15 years after transplantation 107,108 . Post-mortem analysis of the brain of one PD patient transplanted with fVM tissue revealed survival and integration of the graft nearly a quarter-century following transplantation 109 . In some patients, one unforeseen outcome was the development of debilitating graft-induced-dyskinesias—a poorly understood phenomenon 110 –112 . Due to the limited availability and logistical hurdles associated with human fetal tissue, several laboratories have focused on in vitro differentiation 113,114 and transplantation of dopaminergic progenitors using ESCs 114 –116 and iPSCs 117 –119 . The in vitro differentiation protocol of pluripotent stem cells, however, is not a replacement for in vivo development.
Generating authentic nigral dopamine neurons by blastocyst complementation can provide a source of human VM tissue isolated from the fetal chimera, which has been subjected to all the inductive cues of development. In-depth analyses of the development of the substantia nigra have provided a wealth of knowledge on the transcription factors involved and the timing of gene expression in rodents and humans 120 –122 . Identifying a specific gene (or genes) to knock out that will create a specific niche for complementation is difficult. In general, the development of the substantia nigra can be broken down into three stages: regionalization, specification, and maturation. In transgenic mice and chick embryos, knocking out specific genes during the regionalization, specification, or maturation stages has led to varying degrees of cell loss within the substantia nigra. Knockout of Otx2 during the regionalization stage results in significant malformation of the midbrain 123 . Knockout of Nurr1 or Lmx1a during the specification and maturation stage resulted in a reduction of nigral dopaminergic neurons and also resulted in a decrease in ventral tegmental dopaminergic neurons 124 –126 . In the maturation stage, live-born aphakia (Pitx3 mutant) mice present with a nigral specific reduction in the number of dopamine neurons 127,128 .
Oligodendrocyte Progenitors
Oligodendrocytes are the myelinating cells in the central nervous system, aiding in neuronal communication through proper conduction of action potentials. Multiple diseases are associated with oligodendrocyte dysfunction, but four of the most prevalent are multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), traumatic brain injury (TBI), and spinal cord injury (SCI). MS is characterized by the loss of oligodendrocytes resulting in motor deficits, tremors, and visual problems. Currently, there is no cure for MS, and it is a neurodegenerative disease that gradually worsens over time. ALS is characterized primarily by a loss of upper and lower motor neurons, but oligodendrocyte dysfunction largely influences the disease progression through failure to remyelinate and support metabolic function. ALS currently has no cure and results ultimately in death, most commonly from an inability to contract the diaphragm. TBI is the most common cause of morbidity in young people in the United States, with 2,800,000 emergency room visits related to TBI per year 129 . According to the World Health Organization, 250,000–500,000 new SCI cases are reported each year worldwide, resulting in compromised mobility, sensation, and paralysis. Currently, treatments for both TBI and SCI are largely palliative, with no FDA-approved therapies to reverse the damage sustained by the injury.
There are multiple clinical trials attempting to transplant human oligodendrocyte precursor cells (OPCs) into the lesioned CNS. Q Therapeutics has developed a human glial-restricted progenitor cell line with the capability of forming myelinating oligodendrocytes in animal models of MS, ALS, and SCI. Similarly, Asterias Biotherapeutics is attempting to use human ESC-derived OPCs to treat SCI in a phase1/2 clinical trial. Other groups such as Neuralstem, Stem Cells Inc., and FortunaFix have either used, or propose to use, other progenitor cell types such as neural stem cells that are designed to generate OPCs in combination with other neural cell types. All of these groups have published results demonstrating safety and efficacy in rodent models of disease 130 –133 . These studies suggest that OPCs may be an effective therapeutic target for transplantation.
Oligodendrocytes develop initially from the ganglionic eminences of the developing forebrain 134 . These cells begin migrating and differentiating largely after neuronal development from a subset of glial-restricted progenitor cells 134 –136 . For blastocyst complementation, it is important to choose a genetic knockout that is early in OPC development to stall the endogenous population of glial precursor cells before they fill the OPC niches. Conversely, a gene too early in OPC development will likely affect too many developmental pathways, thus potentially leading to undesired cell types in addition to OPCs. Accordingly, the transcription factors Olig1, Olig2, and Sox10 are likely the best candidates to target. Olig2 has been implicated in a number of other cell types, including motor neurons and cholinergic neurons, but still is a relatively specific transcription factor 137,138 . OLIG1 is a later-expressed transcription factor in OPC development, likely initiating the expression of myelination proteins 139 . Additionally, SOX10 is a transcription factor downstream of OLIG2 and has been shown to interact with OLIG1 to initiate myelination 139,140 . Although none of these targets are completely specific to OPCs, they are early enough to ensure that a vacated niche is present. Other target genes include myelin basic protein, oligodendrocyte marker Pdgfrα, or Nkx2.2, although these targets are less promising.
Barriers and Challenges to Complementation
Aside from the ongoing ethical debate surrounding blastocyst complementation, many technical barriers currently need to be addressed. Off-target chimerism with human cells present in the brain or reproductive system of the host animal is one major concern 141 . Conversely, mixed chimerism in the target tissue where animal cells are present within the human organ has also raised concerns over the viability of the transplanted organ, as well as immunologic considerations. All of these issues have been discussed previously 142 –144 . Other technical barriers that need to be addressed include the feasibility of generating genetically engineered embryos, human donor cell characteristics, and the low efficiency of interspecies chimerism.
One major technical problem is the ability to obtain large quantities of genetically engineered large animal blastocysts. In the mouse, where knockout models are abundant, it is possible to obtain fertilized blastocysts carrying the desired genotype from transgenic mouse breeders. As validated and characterized transgenic livestock are in limited supply, it is currently necessary to harvest wild-type oocytes, which can be used to generate parthenogenetic embryos through chemical or electrical activation 145,146 . The uncleaved embryos are then genetically engineered through TALEN or CRISPR injections to inactivate the desired gene 11,147 . This process results in limited knockout blastocysts, as success rate at each step is compounded. To promote survival of the embryos and increase the rate of blastocyst development, anti-apoptotic reagents have been added to the cultures, but there still is no consensus on optimal conditions 148 –150 .
Another technical problem is the chimeric potential of the donor cells. Results from interspecies chimera studies, to date, have not identified the characteristics of a cell line that will best incorporate with the developing embryo. Recent reports suggest that developmental synchronization between host embryo and donor cells appears to be required, with naïve-iPSCs and intermediate-iPSCs contributing to chimeras 5,151 –153 . Methods to enhance the chimeric potential of donor cells have been attempted through inhibiting apoptosis until the developmental stage matches that of the host 151,154 .
Low efficiency of successful interspecies complementation is another critical issue that must be addressed. Theunissen and colleagues reported that 0.2% of human-mouse chimeric embryos implanted into a maternal surrogate developed into a fetus with roughly 1 out of 10,000 cells of human origin 155 . Similarly, less than 0.1% of human-pig chimeric embryos developed into a fetus with roughly 1 out of 100,000 cells of human origin 5 . In a study on intraspecies complementation, 5% of complemented blastocyst resulted in fetuses demonstrating complementation within the pancreas 3 .
Conclusions
The potential of new gene editing technologies, most notably genome modifications using CRISPR and TALENs, is evident from the recent exponential surge in research activity. Use of these technologies in combination with blastocyst complementation could greatly enhance our ability to produce human organs and cells in a relatively short period of time. Furthermore, generation of human-animal organ chimeras together with their associated vasculature could overcome the problems of organ shortage for transplantation, in addition to those associated with immune rejection. However, it will be imperative, for a variety of reasons, that we understand the conditions of stem cell growth in a developmental environment after their injection into a blastocyst, and development of the chimeric tissue. To achieve clinical success in human transplantation using complemented organs produced in animals, we must address critical issues such as species-specific differences in the mechanisms of fetal development, size of developed chimeras for transplantation, the possibility of transmission of zoonotic diseases through human-animal chimeras. The combination of gene editing and blastocyst complementation has created an entirely new and exciting field of medicine—one in which the role of precision medicine becomes a key to long and/or improved quality of life.
Footnotes
Acknowledgment
The authors wish to acknowledge Marra Evans who contributed illustrations for the figures.
Ethical Approval
This study was approved by our institutional review board.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Disclosure
WCL is CSO of Regenevida.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study is supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grant 1R01DK117286-01 to C. J. Steer.