
Abstract
The dynamic and multicellular processes of neuroinflammation are mediated by the nonneuronal cells of the central nervous system, which include astrocytes and the brain’s resident macrophages, microglia. Although initiation of an inflammatory response may be beneficial in response to injury of the nervous system, chronic or maladaptive neuroinflammation can have harmful outcomes in many neurological diseases. An acute neuroinflammatory response is protective when activated neuroglia facilitate tissue repair by releasing anti-inflammatory cytokines and neurotrophic factors. On the other hand, chronic neuroglial activation is a major pathological mechanism in neurodegenerative diseases, likely contributing to neuronal dysfunction, injury, and disease progression. Therefore, the development of specific and sensitive probes for positron emission tomography (PET) studies of neuroinflammation is attracting immense scientific and clinical interest. An early phase of this research emphasized PET studies of the prototypical imaging biomarker of glial activation, translocator protein-18 kDa (TSPO), which presents difficulties for quantitation and lacks absolute cellular specificity. Many alternate molecular targets present themselves for PET imaging of neuroinflammation in vivo, including enzymes, intracellular signaling molecules as well as ionotropic, G-protein coupled, and immunoglobulin receptors. We now review the lead structures in radiotracer development for PET studies of neuroinflammation targets for neurodegenerative diseases extending beyond TSPO, including glycogen synthase kinase 3, monoamine oxidase-B, reactive oxygen species, imidazoline-2 binding sites, cyclooxygenase, the phospholipase A2/arachidonic acid pathway, sphingosine-1-phosphate receptor-1, cannabinoid-2 receptor, the chemokine receptor CX3CR1, purinergic receptors: P2X7 and P2Y12, the receptor for advanced glycation end products, Mer tyrosine kinase, and triggering receptor expressed on myeloid cells-1. We provide a brief overview of the cellular expression and function of these targets, noting their selectivity for astrocytes and/or microglia, and highlight the classes of PET radiotracers that have been investigated in early-stage preclinical or clinical research studies of neuroinflammation.
Keywords
Introduction
Inflammation is an adaptive cellular response triggered by noxious stimuli and conditions, such as infection, tissue injury, or malfunction. 1,2 Famed immunologist Ruslan Medzhitov proposed that inflammation pathways evolved as an adaptive response for restoring homeostasis, as distinct from the chronic and maladaptive parainflammatory response leading to degenerative disease. 3 Initial prohomeostatic adaptive responses engage macrophages and other immune cells to release inflammatory mediators such as cytokines, whereas resolution reinstates the homeostatic state of the tissue, chronic parainflammatory conditions, can entail maladaptive changes in homeostatic set points leading ultimately to degenerative changes.
Neuroinflammation: Relevance to Neurodegenerative Disorders
Neuroinflammation is an inflammatory and adaptive response within the central nervous system (CNS). 4 Neuroinflammation is mediated by the production of small signaling proteins such as cytokines and chemokines, reactive oxygen species (ROS), and lipid second messengers that are produced in situ by glial cells resident in the CNS (ie, astrocytes and microglia), capillary endothelial cells, and immune cells arising in the periphery. These inflammatory factors provoke physiological reactions in relation to the context and duration of the primary stimulus or insult, 4 with self-limiting of the innate immune reactions upon successful resolution of the initiating condition. 5,6 In contrast, chronic neuroinflammation entails persistent activation of microglia and other immune cells in the CNS, with sustained release of inflammatory mediators leading to a feed-forward propagation of the inflammatory response. 7,8
The blood–brain barrier (BBB) maintains the internal metabolic milieu of the brain in conjunction with glial cells, which can exert a concerted inflammatory process in response to stress. 9 Endogenous anti-inflammatory and neuroprotective responses normally keep transient inflammatory reactions in check and return the brain to homeostasis. However, as with chronic inflammatory conditions in peripheral organs, unregulated glial reactions can induce chronic neuroinflammation that promotes or propagates neurodegenerative diseases. 10 -16 For example, in Alzheimer disease (AD), the neuroinflammatory cycle entails sustained activation of microglia and astrocytes in response to stimuli such as amyloid beta (Aβ) deposits. 17 Glia proinflammatory responses activated by Aβ include induction of cytokines (tumor necrosis factor α [TNF]-α, interleukin 1β [IL-1β], and S100 β), chemokines (macrophage inflammatory protein [MIP]-1β), and oxidative stress-related molecules, which can cause neuronal cell dysfunction and/or death and further propagate the inflammatory response. 17 Clinical evidence indicating neuroinflammation as a pharmacological target for chronic neurodegenerative diseases comes from epidemiological observations and immunostaining of postmortem brain tissue. For example, long-term use of nonsteroidal anti-inflammatory drugs (NSAIDs) may impart protection against AD. 18 Although prospective studies did not reveal a protective effect, a cross-sectional follow-up study showed that prior exposure to naproxen was associated with a significant reduction in the incidence of AD, 19 suggesting that NSAIDs may be neuroprotective only at the earliest stages of the AD process. Nonetheless, postmortem brain tissue from patients with AD exhibit overexpression of an array of inflammatory mediators in regions most affected by the primary pathologies, that is, Aβ deposition. 20 Taken together, these observations suggest that neuroinflammation constitutes a viable target for the discovery and development of both diagnostics and therapeutics for AD and, by extension, other neurodegenerative diseases.
Cellular Players of Neuroinflammation: Reactive Astrocytes and Reactive Microglia
Neuroinflammation in diseases such as AD, Parkinson’s disease, and amyotrophic lateral sclerosis manifests in reactive morphology of astrocytes and microglia, accompanied by release of low to moderate levels of inflammatory mediators in the brain parenchyma. We now briefly summarize the key cellular players that regulate neuroinflammatory responses.
Astrocytes are abundant in the gray and white matter of brain, where they modulate neuronal metabolism and ion transit across the BBB. 21 -24 Under pathological conditions, astrocytes undergo a phenotype change from a resting morphology to reactive form with hypertrophic cell bodies and processes. 25 In response to severe injury such as ischemic stroke, reactive astrocytes form a characteristic structure called an astrocyte scar, which prevents infiltration of peripheral inflammatory cells or molecules, thereby reinstating the BBB. 26 -29 Although many reports have shown that reactive astrogliosis is a hallmark neuroprotective reaction of astrocytes against CNS disorders, complex mechanisms underlying reactive astrogliosis and induction of the neuroprotective effects suggest that they can both hinder and support CNS recovery. 25,30,31
Microglia are resident immune cells in the brain, which continuously monitor and respond to the brain microenvironment. 32 Constituting about 10% to 20% of the total glial cells of the brain, microglial expression is highest in the gray matter, including neocortex, hippocampus, olfactory bulb, and the basal ganglia. Upon brain injury, microglia transit from the resting to activation state, which entails a pronounced change from ramified morphology to amoeboid structure with swollen cell bodies and short processes. The amoeboid form is capable of migration to the pathogen site 33 and presumably serves an immune surveillance function. 34,35 Typically, microglia have a very low threshold of activation and rapidly respond within 20 to 40 minutes of injury. 36 Although the causal sequence of microglial activation in neuroinflammatory disorders can be uncertain, the phenotype presents a reliable biomarker for brain injury.
Activated microglia are a double-edged sword, exerting toxic and beneficial roles depending on their polarization phenotype, activation status, and the cellular context. 37,38 Indeed, microglial activation may correspond to the binary activation profile of peripheral monocytes, characterized by M1/M2 phenotypes. 39 The M1-type microglia represent a proinflammatory phenotype releasing inflammatory mediators such as prostaglandins, TNF-α, IL-6, IL-1β, ROS, and glutamate, whereas the M2-type microglia serve to resolve the inflammatory response and express anti-inflammatory mediators such as IL-4, IL-13, IL-25, IL-1ra, insulin-like growth factor 1, and neurotrophic factors. 40
Genetic profiling of reactive astrocytes from mice treated with a systemic injection of lipopolysaccharide (LPS) or upon middle cerebral artery occlusion to induce ischemia reveal 2 different types of reactive astrocytes, namely, A1 and A2, 41 as distinct from the microglia M1/M2 dichotomy noted above. A1 astrocytes upregulate many classical complement cascade genes previously shown to be destructive to synapses, whereas A2 astrocytes upregulate many neurotrophic factors that may restore or protect synaptic integrity. Activated M1 microglia induce the A1 astrocytes, which lose most normal astrocytic functions but gain a new neurotoxic function, rapidly killing neurons and mature, differentiated oligodendrocytes. 42 Importantly, A1-like reactive astrocytes are present in most major neurodegenerative diseases, raising the possibility that they are drivers for neurodegeneration.
Immunohistochemical techniques enable the detection of specific molecular markers and serve as essential tools for identifying and characterizing cells in healthy and pathological tissue. 43 Constitutive expression of ionized calcium-binding adapter molecule-1 (IBA1) is specific for microglia, irrespective of their activation state. 44,45 However, detection of IBA1 in conjunction with M1 or M2 markers can identify the microglial phenotype. 46 Commonly used M1 markers are CD68, a marker of microglial lysosomes indicative of phagocytic microglia, and major histocompatibility complex class II. On the other hand, M2-type microglia express the mannose receptor CD206. 46 Glial fibrillary acid protein (GFAP) is the prototypical marker for immunohistochemical identification of astrocytes. First isolated from postmortem brain of patients with multiple sclerosis (MS), GFAP was immunohistochemically localized within reactive astrocytes. 47,48 Although GFAP is immunohistochemically detectable in many astrocytes throughout the healthy CNS, double staining studies with multiple markers (including transgenic reporter proteins) show that many mature astrocytes in healthy CNS tissue do not express detectable levels of GFAP. Nevertheless, GFAP expression is a sensitive and reliable marker that labels majority of reactive astrocytes responding to CNS injuries. 43 These findings in vitro predict that detection of markers for activated microglia, and astrocytes in vivo should reveal neuroinflammation and associated neurodegenerative disorders.
Positron Emission Tomography Imaging: A Vital Diagnostic Tool for Tracking Neuroinflammatory Biomarkers and Diagnosing Neurodegenerative Diseases
In recent years, technical advances in imaging modalities such as positron emission tomography (PET) and magnetic resonance imaging (MRI) have encouraged their use for the evaluation of functional, neurochemical, and anatomical changes in the diseased brain. 49 PET is a noninvasive molecular imaging technique that enables quantification and visualization of physiological processes by recording the time-dependent distribution in living organs of tracer molecules labeled with positron-emitting isotopes. Development of novel radiotracers targeting diverse receptors, transporters, enzymes, and other molecular targets within the human brain has expanded the use of PET neuroimaging, specifically in neurodegenerative diseases, and has been reviewed by several groups. 50 -57 Among its clinical research applications, PET can quantify neuropathological markers or reveal pharmacokinetic and pharmacodynamic properties of a drug candidate. Thus, it is not surprising that CNS PET imaging has become central for establishing proof of mechanism and optimal dosing for novel therapeutic agents, thereby allowing accelerated decision-making in clinical trials.
Nonetheless, developing radioligands for PET imaging of molecular targets in the CNS is a demanding task, not least of all due to aspects of quantitation. In the first instance, a candidate tracer must cross the BBB, which can be an obstacle due to binding to plasma proteins, low intrinsic permeability, or active extrusion from the brain. In general, passive transfer across the BBB is promoted by low molecular weight <500 Da, low hydrogen-binding capability, lack of formal charge, and moderate lipophilicity (Log D range = 2.0-3.5). 58,59 Tissue uptake of PET tracers is often assessed as percentage of injected dose per gram of target tissue (standard uptake volume [SUV]). A peak uptake (SUVpeak) <2% ID/g generally indicates sufficient brain exposure for quantitation measurement of specific binding. Upon entering the CNS, an effective radiotracer must bind with sufficient affinity and specificity to its target binding site, which must be of adequate abundance to be detectable against a background of nonspecific binding. For reversibly binding tracers, specific binding will increase as a function of time until attaining an equilibrium defined by B max, the concentration of the binding site, and K D, the affinity of interaction between the tracer and its binding site, 60 where the ratio B max/K D represents binding potential (BPND). In practice, quantitation is difficult when BPND <1 due to low specific signal and when BPND >10 due to failure to attain equilibrium within the time constraints of a dynamic PET scan. 58,61 However, very high BPND can be an advantage for semiquantitative evaluation of diagnostic radiotracers in a clinical setting. 62 An ideal PET radiotracer to image neuroinflammation should not only have an adequate signal-to-noise ratio (specific vs nonspecific binding) but should also yield a good pathological versus baseline (normal) signal that is indicative of an inflammatory process. However, clinical translation of candidate radiotracer can fail due to deficiencies in animal models, which may not emulate key features of human neurodegenerative diseases. 63 -65 The first phase of molecular imaging of neuroinflammation was led by PET studies with isoquinoline carboxamide [11C]PK11195, the prototype ligand of translocator protein-18 kDa (TSPO), formerly known as the peripheral benzodiazepine site. 66,67 The TSPO is a highly hydrophobic 5 transmembrane domain protein expressed in the outer mitochondrial membrane of microglia and other cells of macrophage lineage. 68 The TSPO is thought to mediate the transport of cholesterol into the inner compartments of mitochondria. While constitutively expressed in healthy brain, microglial TSPO expression is substantially upregulated in response to brain injury and in several neuroinflammatory diseases, 69,70 although there is also TSPO expression in reactive astrocytes. 71 Despite this ambiguity about its cellular origin, immense clinical interest has emerged in TSPO imaging as a biomarker for neuroinflammation in neurodegenerative diseases. We have recently summarized the binding properties as well as challenges for quantitation and interpretation PET studies of [11C]PK11195 versus a dozen second- and third-generation TSPO tracers, including [18F]FEDAA1106, [11C]PBR28, [18F]PBR06, [11C]DPA-713, [11C]ER176, and [18F]DPA-714, belonging to 5 structural classes. 72 The new TSPO tracers generally have 2- to 4-fold higher BPND than [11C]PK11195 in healthy brain, with the caveat that their binding affinity is often subject to a common TSPO allelic variant in human populations. Consequently, recent efforts have focused on developing high-binding TSPO radiotracers that are insensitive to genotype, thus obviating the need to stratify patients by allelic status. 73 Furthermore, available TSPO ligands do not distinguish between pro- and anti-inflammatory microglia, which might be differentially expressed at the onset and progression of a neuroinflammatory condition. 74 In any event, TSPO is just one aspect of inflammatory responses, and there is a need to develop PET tracers targeting specific biochemical markers of specific cell types involved in neuroinflammation; this is the focus of the present review.
Emerging Targets and Tracers for PET Imaging of Neuroinflammatory Biomarkers
Molecular targets for neuroinflammation imaging discussed herein can be broadly categorized into (1) enzymes and intracellular signaling molecules including glycogen synthase kinase 3 (GSK-3), monoamine oxidase-B (MAO-B), ROS, imidazoline-2 binding sites (I2BS), cyclooxygenase (COX), and arachidonic acid; (2) G-protein coupled and ionotropic receptors such as sphingosine-1-phosphate receptor 1 (S1P1), cannabinoid-2 receptor (CB2), the chemokine receptor CX3CR1, and the P2X7 and P2Y12 purinergic receptors; and (3) members of the immunoglobulin receptor superfamily, including the receptor for advanced glycation end products (RAGE), Mer tyrosine kinase (MerTK), and triggering receptor expressed on myeloid cells-1 (TREM1). We present an overview of (1) the cellular expression and function of these targets, (2) their selectivity for microglia and/or astrocytes, and (3) PET radiotracers for the respective targets that have been investigated in preclinical models of neuroinflammation. The molecular targets are listed in Table 1, and the structures of corresponding lead PET radiotracers are shown in Figure 1.
PET Imaging of Neuroinflammation: Molecular Targets and Potential Radiotracers.

Abbreviations: CB2: cannabinoid-2 receptor; COX: cyclooxygenase; GSK-3: glycogen synthase kinase 3; I2BS: imidazoline-2 binding sites; MAO-B: monoamine oxidase-B; MerTK: Mer tyrosine kinase; RAGE: the receptor for advanced glycation end products; ROS: reactive oxygen species; S1P1: sphingosine-1-phosphate receptor 1; TSPO: translocator protein-18 kDa; TREM1: triggering receptor expressed on myeloid cells-1.
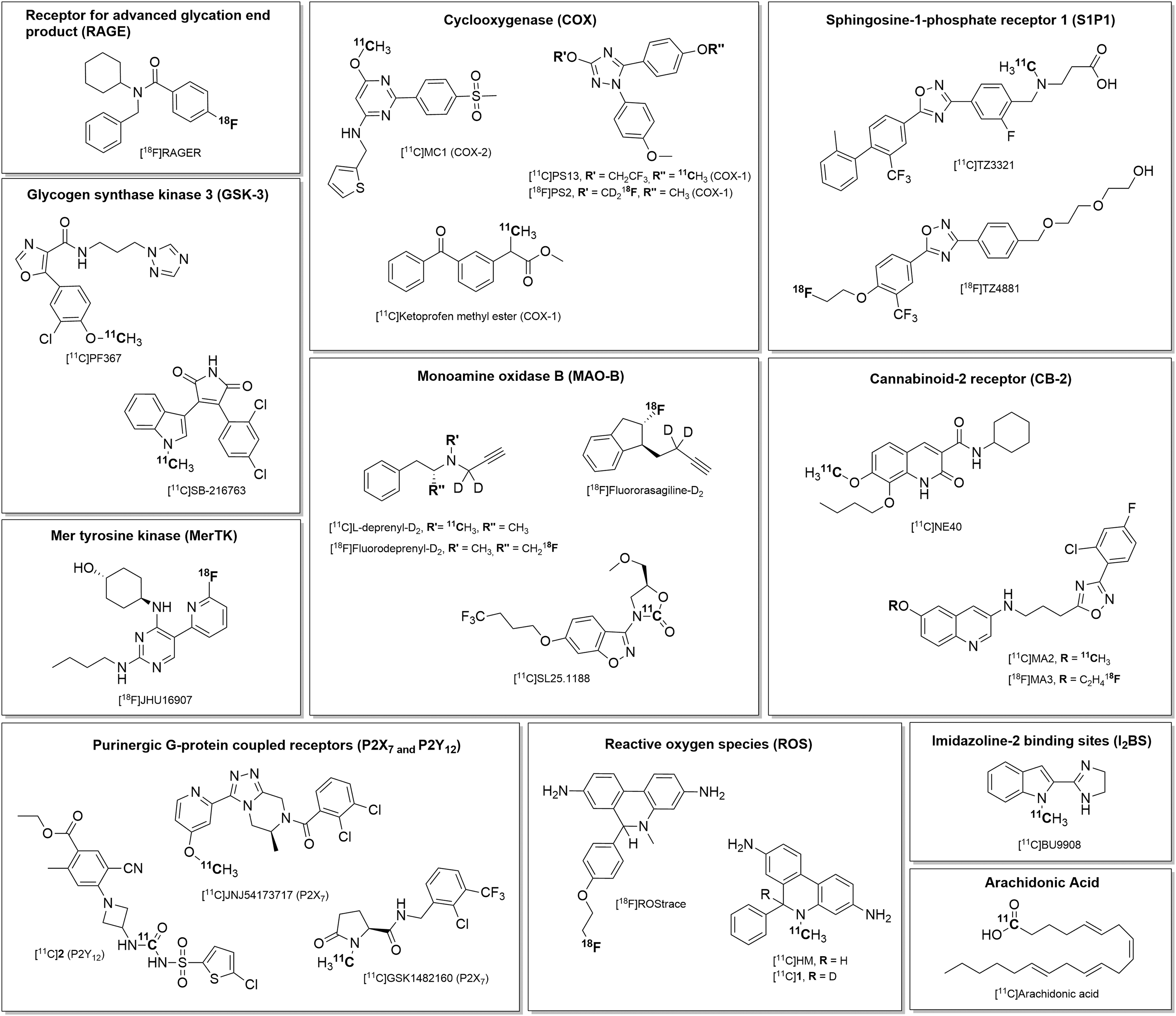
Chemical structures of selected PET radiotracers for PET imaging of neuroinflammation. PET indicates positron emission tomography.
Glycogen Synthase Kinase 3
Glycogen synthase kinase 3 is a serine/threonine kinase that exists in 2 closely related isoforms, namely, GSK-3α and GSK-3β. 75,76 While GSK-3α primarily regulates glycogen storage in the liver, the GSK-3β isoform is highly expressed in neural tissue, where it regulates brain development. 77,78 In rat cerebral cortex, GSK-3β is expressed in both astrocytes and microglia and is activated following exposure to LPS. 79 However, both messenger RNA (mRNA) isoforms are abundantly expressed in hippocampal and cortical neurons and in Purkinje cells. The GSK-3 regulates various complex biological processes, including glucose metabolism, cell signaling, cellular transport, apoptosis, proliferation, and intracellular communication. 80 -82 Experimental autoimmune encephalomyelitis (EAE) is a T-helper cell-mediated autoimmune disease characterized by T cell and monocyte infiltration in the CNS associated with local inflammation. 83 Given that EAE leads to an approximation of the key clinical, immunological, and neuropathological features of MS, the rodent EAE model has been widely adopted for preclinical investigation of therapeutic targets and evaluation of drug candidates for MS. 83,84 Compared to wild-type mice, constitutively active GSK-3α/β knock-in mice had more rapidly progressing and severe EAE. 84 Furthermore, postmortem studies show increased GSK-3β activity in postmortem brain samples from patients with AD. 85 GSK-3 inhibition may, therefore, present a rational strategy for treating neuroinflammatory diseases, and GSK-3 PET could be used to measure inhibition by GSK-3 therapeutics. Given the role of GSK-3β in phosphorylation of tau and Aβ production, 86 in vivo imaging of GSK-3 might open a path to detecting increased tau hyperphosphorylation, Aβ aggregation, and glia-mediated inflammatory responses, all of which are hallmarks of AD and non-AD tauopathies.
Given the wide range of functions associated with GSK-3, it is unsurprising that GSK-3 has emerged as a target for CNS drug development and medical imaging in various disease settings. 53 -55,87 For neurodegenerative diseases, GSK-3 PET imaging could indicate target engagement by GSK-3 therapeutics and offer a path to diagnostic agents that correlate with not only early cognitive impairment of AD but also increased tau hyperphosphorylation, 88,89 increased Aβ production, and local plaque-associated glial-mediated inflammatory responses, 90,91 all of which are hallmarks of AD and non-AD tauopathies. The greatest obstacle for molecular neuroimaging of GSK-3 has been to identify potent and highly selective small molecular weight ligands with sufficient brain penetration. We have developed the PET radiotracer for GSK-3 focusing on the synthesis of 11C-labeled isotopologues of AR-A014418, and other laboratories have subsequently explored different scaffolds. 53,92 Only a handful of radiotracers for GSK-3 have been studied in vivo. Among these, [11C]SB-216763 showed good brain uptake in rodents and nonhuman primates (NHPs) but was not selective for GSK-3 among structurally similar kinases. 93,94 [11C]PyrATP-1- and 11C-oxadiazole-based radiotracers failed to show appreciable uptake in vivo. 95,96 We also explored 18F-labeled maleimide-based tracers for GSK-3. 97 Although preliminary PET imaging with these compounds showed moderate uptake, we saw no displaceable binding in rodent brain.
In collaboration with Pfizer, our team recently discovered PF-367, one of the most potent and selective inhibitors of GSK-3 reported to date, with proven efficacy in vitro and in vivo efficacy in modulating tau phosphorylation. 98 [11C]PF-367 readily crosses the BBB in NHP (SUVpeak = 1 at 5 minutes postinjection). Pharmacological blocking provoked relatively rapid washout of [11C]PF-367 from the brain, indicating a high-binding signal. Due to its high BBB permeability and admirable selectivity for GSK-3 in a panel of protein kinases, [11C]PF-367 represents a lead neuroimaging agent for GSK-3.
Monoamine Oxidase-B
Monoamine oxidase-B is a flavin–adenosine–dinucleotide enzyme of the outer mitochondrial membrane enzyme that catalyzes the oxidative deamination of dopamine and β-phenylethylamine but with lesser affinity for serotonin, noradrenaline, and tyramine; the suicide inhibitors pargyline and L-deprenyl evoke irreversible MAO-B inhibition. 99,100 The highest expression of the MAO-B gene in human brain is in the frontal cortex and locus coeruleus, but histological examination reveals focally high activities of MAO-B within serotonin and histamine neurons of murine brain. 101,102 Nonetheless, the great bulk of brain MAO-B apparently resides in astrocytes as revealed by microautoradiography with L-[3H]deprenyl. 103 Furthermore, that study showed increased expression of MAO-B in spinal cord from patients diagnosed with motor neuron disease, which colocalized in reactive astrocytes double labeled for GFAP. During its catalytic cycle, MAO-B, like all amine oxidases, produces hydrogen peroxide, which may give rise to other ROS 104 in idiopathic neurodegenerative diseases. Increased astrocytic MAO-B activity due to astrocytosis in brain of patients with AD may impart oxidative stress, further exacerbating the ongoing neurodegeneration. 105,106 Therefore, MAO-B inhibition might be neuroprotective in diseases where ROS generation is implicated in neurodegeneration. 107
Several radiotracers have been developed for MAO-B PET imaging, yet the majority of clinical research studies have been led using the propargyl compound L-[11C]deprenyl to evaluate MAO-B in human neuropsychiatric studies. 54,108,109 In the past decade, several radiolabeled analogs of L-deprenyl (selegiline) have been developed and investigated for detection of reactive astrogliosis in neuroinflammation disorders. In autoradiographic studies with [11C]-L-deprenyl, there was significantly higher binding in temporal lobe and the white matter of patients with AD compared to controls. 106 Moreover, increased regional radioligand binding was accompanied by an increased number of activated astrocytes as demonstrated by GFAP immunohistochemistry in adjacent brain slices. While the MAO-B suicide inhibitors L-deprenyl and rasagiline effectively blocked [11C]-L-deprenyl binding in vitro, the MAO-A inhibitor pirlindole had not such effect.106 The PET studies with [18F]fluorodeprenyl showed highest specific binding in striatum, intermediate uptake in thalamus and cortex, and lowest binding in cerebellum of NHP. 110 However, the irreversible binding mechanism of the “suicide substrate” propargyl compounds can hinder quantitation in PET studies, since the trapping rate approaches the limit established by perfusion/delivery of the tracer, which led to development of the isotopologs L-[11C]deprenyl-D2 and [18F]fluorodeprenyl-D2. These compounds gave improved MAO-B quantitation through the deuterium isotope effect, which slowed the rate of irreversible binding to MAO-B. 109,111 Indeed, PET studies with [18F]fluorodeprenyl-D2 revealed favorable kinetic properties with relatively fast washout from NHP brain and improved sensitivity for MAO-B imaging, which was recapitulated in a comparison of [18F]fluororasagiline-D2 versus [18F]fluororasagiline. 112 Nonetheless, the potentially brain-penetrating radioactive metabolites of propargyl compounds are problematic for quantitation.
An alternative approach for measuring MAO activity in vivo is the use of metabolic trapping, where the product of the catalytic activity of the enzyme is not covalently bound but is still retained within the target tissue. 113 To that end, [11C]1-methyl-4-phenoxy-1,2,3,6-tetrahydropyridine ([11C]PHXY) and [11C]4-methyl-7-(pyridin-4-yloxy)-2H-chromen-2-one ([11C]COU)1-[11C]methyl-4-aryloxy-1,2,3,6-tetrahydropyridines were investigated as metabolic trapping agents for monoamine oxidases using micro-PET imaging in rat and NHP brain. 113 Isozyme selectivity was determined after pretreatments with deprenyl (selective MAO-B inhibitor) or clorgyline (selective MAO-A inhibitor) or both irreversible inhibitors prior to scan. While both radiotracers evaluated failed to exhibit specificity for either isozyme in the rat brain, in vivo trapping of [11C]PHXY was more sensitive to MAO-A inhibition, and [11C]COU was more sensitive to MAO-B inhibition. Given the encouraging isozyme selectivity of [11C]PHXY and [11C]COU in the monkey brain, further evaluation of these radiotracers will be beneficial.
Our laboratory has been working over the past decade to develop several 11C- and 18F-labeled PET radiotracers for imaging MAO-B based on coumarin, pyrrole, and propargylamine scaffolds in preclinical studies. 114 A reversible MAO-B ligand [11C]SL25.1188 115 has been synthesized by us via a novel [11C]CO2 fixation technology and validated for human use. 116 Preclinical characterization in NHP revealed very slow plasma metabolism, high brain uptake, and reversible binding kinetics in brain. 117 We and our collaborators have recently successfully translated [11C]SL2511.88 for first-in-human clinical research studies, validating it as the first reversibly binding MAO-B radiotracer for human use. 118 It is presently being explored in several patient populations in clinical PET research studies, and we are working toward development of a fluorine-labeled derivative of SL25.1188. Our initial array of 18F-labeled compounds showed fast binding kinetics and specificity to MAO-B ex vivo but imparted excessive cranial uptake due to defluorination in vivo. 119 Further optimization and evaluation of reversible MAO-B tracers in neuroinflammation are underway.
Aggregation and hyperphosphorylation of tau protein leading to the formation of pathological tau deposits are the hallmark of several neurodegenerative diseases including AD. Advances of molecular imaging in the recent years have led to the development of promising tau-PET tracers such as THK5317, THK5351, AV-1451, PBB3, and MK-6240. 120 Interestingly, off-target binding of tau-PET tracers are potentially attributed to MAO-A and/or MAO-B enzymes. 121 For example, binding to MAO-B in vivo in humans was recently demonstrated for another tau-PET tracer, [18F]THK5351, in which selegiline (MAO-B inhibitor) reduced the cortical PET signal by 40% in patients with AD. 122 Taken together, tau-PET tracers devoid of affinity for MAO-A and MAO-B will need to be developed for diagnosis and monitoring of disease progression or treatment efficacy.
Reactive Oxygen Species
Oxidative stress is produced by various enzymatic reactions and chemical processes when ROS generation exceeds the capacity of antioxidant defense mechanisms, which include a network of compartmentalized antioxidant enzymic and nonenzymic molecules usually distributed within the cytoplasm and various cell organelles. 123 The ROS particularly implicated in oxidative stress include superoxide anion (O2 ·−), hydrogen peroxide (H2O2), and hydroxyl radical (·OH). 124,125 Under normal physiological conditions, microglial cells (like all living cells) express low ROS levels due to tight regulation by antioxidant pathways. 126 Some basal expression of oxidative stress molecules is essential for many physiological functions such as cell survival, proliferation, differentiation, and apoptosis. 123 After an inflammatory challenge, microglial cells differentiate into the activated M1 phenotype that evoke a rapid increase in ROS levels, with positive feedback further upregulating the proinflammatory M1 gene profile. Although this may be an advantageous acute cellular response, excessive and sustained ROS production may be responsible for tissue injury in neurodegenerative diseases with an inflammatory component. Uncontrolled ROS production can damage DNA and react with lipids, proteins, and carbohydrates, thus impairing cellular functions while further exacerbating inflammatory reactions. 123 Several reports have shown that cerebral ROS levels tend to increase with age, perhaps in conjunction with increased MAO-B activity. Moreover, elevated ROS levels are detected in the brain of patients with AD as well as cellular and animal models of AD. 127 Overproduction of ROS is considered a contributor to the degeneration of nigrostriatal dopamine neurons in Parkinson disease, 128 as noted in “Monoamine Oxidase-B” section.
A new PET radiotracer, [11C]ascorbic acid, exhibits potential to measure ROS-dependent cellular accumulation. 129 [11C]ascorbic acid was applied to a model of endogenous ROS production, namely, stimulated neutrophil-lineage cells undergoing oxidative burst. For this study, HL60 human promeylocyctic leukemia cells and freshly isolated human neutrophils were employed. During phagocytosis, neutrophils undergo Nox-mediated generation of ROS to destroy bacteria and simultaneously oxidize extracellular ascorbic acid. To investigate tracer uptake via this mechanism, cells were incubated with 10 µCi [11C]ascorbic acid in the presence or absence of phorbol 12-myristate 13-acetate (2 µmol/L) and blocking agent, cytochalasin B (20 µg/mL). 129 For both HL60 cells and neutrophils, a significant increase in cell-associated activity and approximately 2-fold increase in tracer uptake was noted with activation. These results suggest the potential role of [11C]ascorbic acid, more specifically the ascorbate recycling mechanism, to image ROS-driven neuroinflammatory disease states.
Recent studies report on 2 potential radiotracers for PET imaging of ROS. Hydromethidine (HM) is the closely related analogue of hydroethidine that has been studied extensively as a fluorescent probe for the detection of ROS.
130
The underlying hypothesis is that HM, being neutral, readily crosses cell membranes including those of the BBB, while oxidized species are trapped due to their charge and/or by interchelation with cellular DNA.
130
In one report, [11C]HM and its deuterated isotopolog, [11C]

PET image of rat brain with [11C]HM following unilateral intrastriatal (right) injection of sodium nitroprusside (saline in left striatum). 131 (A) Average PET images (0-60 minutes, transverse, coronal, and sagittal views) are aligned with MRI (the surgery sites; * are indicated in the transverse and sagittal MRI), with cylindrical ROS placed in right and left striatum and right and left cerebellum; (B, top panel) time–activity curves (TACs) between right and left side striatum or cerebellum and whole brain; (B, bottom panel) SUV ratio of right to left striatum increased linearly with time, whereas SUV ratio of right to left cerebellum displayed no significant change with time. PET indicates positron emission tomography; MRI, magnetic resonance imaging; ROS, reactive oxygen species; SUV, standard uptake value.
Imidazoline-2 Binding Sites
The imidazoline derivative idazoxan was originally developed as a selective antagonist for α2-adrenoceptor. 133 However, binding studies showed that [3H]idazoxan had affinity for nonadrenoceptor sites that were termed imidazoline-binding sites or imidazoline-preferring receptors. 134,135 Although the endogenous ligand is unknown, the idazoxan-preferring subtype, I2BS, is expressed in several tissues, including the brain, where it is located on astroglial cells and seem to be involved in the regulation of GFAP expression. 135 At a subcellular level, I2BS is also expressed in mitochondrial membranes of astrocytes. The density of I2BS-binding sites is elevated in AD brain postmortem. Alterations in I2BS density have been seen in neurodegenerative diseases as evidenced by immunohistochemical studies conducted on human postmortem brains from patients with AD and Huntington disease. 135,136 In cortical membranes of patients with AD, I2BS density ([3H]idazoxan binding) was markedly increased (63%) in comparison to control patients. By contrast, I2BS density was markedly decreased (56%) in the putamen of patients with Huntington disease. In addition, I2BS is a potential imaging marker for human glioblastomas. 137 Increased I2BS density was found in glial astrocytic tumors than in normal healthy brain specimens as evidenced by [3H]idazoxan-binding studies conducted in surgical specimens of human glial tumors and normal healthy tissues. 137 Moreover, enhanced I2BS density was more specific for glial than nonglial tumors, suggesting the potential role of PET tracers for I2BS as a potential imaging marker for human glioblastomas.
Recent efforts directed toward elucidation of the role of I2BS in disease states inspired the development of [11C]BU99008, a PET radiotracer for I2BS imaging. 138 In vitro characterization of BU99008 demonstrated its selectivity and nanomolar affinity for I2BS in rodent and cynomolgus brain. Further, in vitro competition studies with the selective I2BS blocker BU224 against [3H]BU99008 binding revealed a 2-site fit in membranes from rat brain, whereas BU224 displacement indicated a single-site fit in cynomolgus brain. BU99008 apparently has affinity for a second binding site in rodent brain, unrelated to I2BS. On structural grounds, BU99008 might be expected to have off-target binding to MAO, and indeed the I2BS is recognized to be a modulatory site present in a subpopulation of MAO-B enzymes. 139 While MAO-A and MAO-B inhibitors evoked little displacement of I2BS-binding sites in cynomolgus brain membrane preparations, coincubation with an MAO-B inhibitor displaced the lower affinity binding component of [3H]BU99008 in rodent brain. 138 Despite the higher MAO-B activity in NHP compared to rodents, [3H]BU99008 lacked significant off-target binding to MAO-B in cynomolgus brain membranes, suggesting a species difference in the off-target binding.
In vivo evaluation of [11C]BU99008 showed rapid uptake in the NHP brain, with highest uptake in the globus pallidus, caudate, and thalamus; moderate uptake in the putamen and parts of cerebral cortex (cingulate, frontal, and insula); and lowest uptake in the cerebellum and occipital cortex. 138 In vivo competition studies revealed that [11C]BU99008 displayed reversible binding kinetics and specificity for the I2BS site. Importantly, in vivo blocking studies using the MAO-A and MAO-B inhibitors, moclobemide and lazabemide, respectively, did not cause any significant change in binding signal of [11C]BU99008. 138 A recent comprehensive PET imaging study of [11C]BU99008 in rats showed great sensitivity to minor and focal changes in I2BS expression. 140 The specificity of this tracer for the I2BS site in rat brain remains to be investigated by MAO displacement studies. While relative expression levels of I2BS and MAO differ from species to species, human brain uptake and in vivo binding characteristics of [11C]BU99008 seem likely to match that in NHP brain. [11C]BU99008 may thus prove useful for elucidating new functions of I2BS in human neuroinflammation and neurodegenerative disorders, 140 although further preclinical evaluation seems warranted to address questions with respect to radioligand specificity for this target. In addition, selective radioligands could aid in the neurochemical search for the endogenous I2BS ligand.
Cyclooxygenase
Cyclooxygenase is the key rate-limiting enzyme in the conversion of arachidonic acid to prostaglandins, which are lipid mediators involved in several physiological and pathological processes including inflammation. 141 The 2 distinct COX isoforms, namely, COX-1 and COX-2, have differing tissue distribution and regulatory mechanisms, although both are blocked by aspirin and ibuprofen. The COX-1 is a constitutively expressed “housekeeping” enzyme present in most tissues, which is primarily responsible for homeostatic prostaglandin synthesis. 142 The COX-2 is mainly induced in response to inflammatory stimuli, although COX-2 is also constitutively expressed in hippocampal and cortical glutamatergic neurons, where it regulates synaptic activity and long-term synaptic plasticity. 143,144
In the CNS, COX-1 immunoreactivity was shown to be enriched in midbrain, pons, and medulla. 145 The COX-1 has recently emerged as a prominent player in neuroinflammation, and COX-1–expressing activated microglia are found around Aβ plaques in human AD brain. 146 Moreover, a selective increase in COX-1 mRNA expression was reported in the hippocampus of aged rats, highlighting the broad relationship between neuroinflammation and aging. 147 The COX-1–deficient mice exhibit decreased inflammatory response, leukocyte infiltration, oxidative stress, and neuronal damage after central injection of the proinflammatory compounds LPS or Aβ1–42. 148,149 Consistent with these findings, treatment of 20-month-old triple transgenic AD mice with the COX-1 selective inhibitor SC-560, improved spatial learning and memory, reduced Aβ deposits and tau hyperphosphorylation, attenuated glial activation and brain expression of inflammatory markers, and switched the activated microglia to an M2 phenotype with enhanced phagocytic ability. 150 In a mouse model of chronic neuroinflammation due to IL-1β overexpression, COX-1 and prostaglandin E2 were upregulated, but this effect was completely abrogated upon genetic deletion or pharmacological inhibition of COX-1. 151 Thus, COX-1 activation is a mediator in acute and chronic neuroinflammatory conditions.
In contrast to COX-1, there are conflicting views of the role of COX-2 in neuroinflammation. The COX-2 inhibition or genetic deletion has been linked to detrimental effects, suggesting a neuroprotective role of COX-2. Thus, COX-2-null mice show increased glial activation and inflammatory markers after LPS injection compared to wild type. 152 The expression of COX-2 changes across disease stages in AD, with early upregulation and late downregulation, in a manner tracking the prostaglandin E2 levels in cerebrospinal fluid. 153,154
The clear role of COX-1 in neuroinflammation has motivated the search for a selective COX-1 imaging probe. 11C-labeled arylpropionic acids and their methyl esters, including [11C]ibuprofen and [11C]ketoprofen-methyl ester ([11C]KTP-Me), have been prepared as potential PET tracers for COX-1. 155,156 Increased plasma protein binding associated with ibuprofen and relatively lower potency for COX-1 compared to KTP-Me (IC50: 7.6 vs 0.047 μmol/L, respectively) likely discouraged further pursuit of [11C]ibuprofen for PET imaging studies. 157,158 [11C]KTP-Me was the first successful PET radioligand for COX-1 in activated microglia. 156 Ex vivo autoradiographic analysis revealed a significant reduction in [11C]KTP-Me accumulation only in the hippocampus, cerebral cortex, and cerebellum of COX-1–deficient mice, but not COX-2–deficient mice, when compared to respective wild-type controls, 156 providing evidence that [11C]KTP-Me selectively binds to COX-1 in the brain. PET imaging with [11C]KTP-Me in rats demonstrated good brain uptake and retention in microglia of inflammatory lesions formed after intrastriatal LPS injection. The first-in-human exploratory study with this tracer revealed favorable biodistribution. 159 [11C]KTP-Me readily entered the human brain, attaining an SUVpeak of 1.5 in cortex at 2 minutes postinjection, followed by a slow washout to 40% of peak activity at 60 minutes. 159 Analysis of plasma extracts showed rapid metabolism of circulating [11C]KTP-Me to [11C]KTP, which may confound analysis of the brain PET signal.
Recent studies report the in vivo evaluation of novel and selective PET radioligands for COX-1 in rhesus monkeys. 160,161 A 11C-labeled radiotracer was developed from PS13 (1,5-bis(4-methoxyphenyl)-3-(2,2,2-trifluoroethoxy)-1H -1,2,4 trazole) that exhibited high affinity and 1000-fold selectivity for NHP and human COX-1 (IC50 ∼1 nM) versus COX-2 (IC50 >1 μmol/L). 160 In vivo PET studies conducted in monkeys demonstrated high brain uptake after [11C]PS13 injection (SUVpeak = 4.4), which was substantially displaced by pretreatment with COX-1 inhibitors (PS13 or KTP-Me). Most importantly, higher radioactivity uptake was found ipsilateral to intracerebral injections of an inflammogen (LPS) or excitotoxin (ibotenic acid) compared to the untreated contralateral side. A [18F]fluoro-D2-methoxy analog ([18F]PS2), with a longer plasma half-life due to the deuterium isotope effect, showing high affinity for human COX-1 (IC50 = 4 nM) and high selectivity (>200-fold) over COX-2, was evaluated in vivo in monkeys. 161 The PET studies in rhesus monkey revealed rapid brain uptake following injection of [18F]PS2 (SUV ∼4.5 at 2 minutes). Moreover, the specific binding of [18F]PS2 to COX-1 was reduced in a self-blocking experiment, supporting its use in further investigations.
A novel and selective ligand MC1 (6-methoxy-2-(4-(methylsulfonyl)phenyl)-N-(thiophen-2-ylmethyl)pyrimidin-4-amine) showed high affinity (IC50 = 3 nmol/L) and selectivity (>3000-fold over COX-1) for COX-2, and its PET analog [11C]MC1 was evaluated in rhesus monkeys. 162,163 Dynamic PET imaging showed an SUVpeak (2.9) at 2 minutes postinjection of the radiotracer, which was blocked by pretreatment with nonradioactive MC1. 162 As a follow-up to this initial study, the same group evaluated brain uptake of [11C]MC1 in monkeys with neuroinflammation due to intracerebral LPS injection 163 ; this treatment increased the displaceable [11C]MC1 uptake at the injection suite. Overall, [11C]PS13, [18F]PS2, and [11C]MC1 are promising tracers for molecular imaging of COX-1 and COX-2 in neuroinflammation.
Arachidonic Acid
Arachidonic acid is a polyunsaturated omega-6 fatty acid derived from phospholipids by neuroreceptor-mediated activation of phospholipase A2 (PLA2) enzyme. 164 Upon mobilization into the intracellular space, arachidonic acid acts as a lipid second messenger for the regulation of signaling enzymes, such as other phospholipases and protein kinase C, while also serving as a precursor for the biosynthesis of prostaglandins and leukotrienes. Its liberation from phospholipids creates a sink in vivo for incorporation of [11C]arachidonic acid into cell membranes such that the PET signal with [11C]arachidonic acid indicates the activity of PLA2 in living brain. 165 The tracer uptake may occur in postsynaptic neuronal elements, and in astroglia and microglia, which likewise express PLA2. However, inflammatory signals arising from activated microglia can stimulate PLA2 via receptor-mediated effects of glutamate or cytokines such that [11C]arachidonic acid can be an indirect index of microglial activation. Thus, increased cerebral [11C]arachidonic acid uptake is taken to indicate a chronic inflammatory condition in patients with AD. 166
Sphingosine-1-Phosphate Receptor 1
The S1P is a bioactive sphingolipid metabolite that regulates critical cellular processes including proliferation, survival, and migration. 167 It is derived from sphingosine upon adenosine triphosphate (ATP)-dependent phosphorylation by sphingosine kinase (predominantly sphingosine kinase 1). S1P is interconvertible with ceramide, a critical mediator of apoptosis, and the prevailing ratio between S1P and ceramide intracellular concentrations is postulated to determine cell fate, as the sphingolipid metabolites have opposing effects on cell survival. 167 Although there are proposed intracellular signaling pathways for S1P, it is extensively extruded via specific transporters from the intracellular to the extracellular environment, where it acts in an autocrine or paracrine manner on S1P receptors. 168,169 Data from in vitro studies and animal models suggest that S1P is important for several physiological CNS functions, including (1) migration of neuronal progenitor cells toward areas of damage; (2) migration of astrocytes and communication of astrocytes with other CNS cells; (3) the regulation of oligodendrocyte survival, function, and modulation of myelination following injury; (4) regulation of microglial number and activation; and (5) maintenance of BBB integrity. 170 -175 Signals initiated by S1P are transduced by 5 G-protein–coupled receptors known as S1P1-5. 176 The understanding of S1P receptor function and biology has advanced rapidly, and these receptors have emerged as attractive therapeutic targets in chronic inflammatory pathologies, autoimmunity, and cancer. Herein, we particularly focus on the role of S1P1 receptors in the context of neuroinflammation and their potential as targets for PET radiotracer development.
The S1P1 receptors are widely distributed within the immune system and CNS. In the CNS, S1P1 is predominantly expressed in neurons and microglia and, to a relatively lesser extent, in astrocytes and oligodendrocytes. 177 The SIP analog fingolimod (FTY720) is an immune-modifying drug derived from the natural product, myriocin. 178 Specifically, FTY720 acts as a functional S1P1 receptor antagonist with anti-inflammatory effects mediated through receptors expressed on lymphocytes, thereby preventing their transit from peripheral lymphoid organs into the CNS. The S1P1 receptor is highly expressed under neuroinflammatory conditions and particularly in MS lesions. 177 In 2010, the US Food and Drug Administration approved FTY720 as the first oral disease-modifying drug to treat relapses of MS. 179 In addition to blocking lymphocyte migration, FTY720 readily crosses the BBB and may attenuate neuroinflammation by regulating the activation and neuroprotective effects of microglia mainly via S1P1 blockade. 180 -182 In vitro studies indicated that FTY720 downregulates production of proinflammatory molecules by microglia while increasing neurotrophic factor production, resulting in an overall neuroprotective phenotype. 182 FTY720 also inhibited secretory vesicle mobility and exocytic release by astroglia, thus attenuating the release of proinflammatory mediators. 183 In support of this finding, in vitro treatment of a human astrocyte cell line with FTY720 suppressed S1P-induced production of proinflammatory cytokines. 184
The EAE is a well-established preclinical model of MS pathology. 185 Specific deletion of the S1P1 receptor in astrocytes resulted in decreased EAE pathology and a loss of FTY720 efficacy, indicating that the primary target of FTY720 during EAE was S1P1 receptors expressed specifically on astrocytes. 186 A potential therapeutic role of FTY720 in AD is also highlighted in a study revealing FTY720 treatment to decrease Aβ plaque density, activated microglia expression, and immunoreactivity of the astrocyte marker GFAP in an AD mouse model. 187 Overall, the discovery of FTY720 has advanced the understanding of physiological actions of S1P receptors and served as a pharmacological tool to identify their role in neuroinflammatory processes and diseases. Moreover, approval of FTY720 for MS treatment has accelerated drug discovery in this field and promoted the development of selective S1P1 agonists (ie, AUY954, CS-0777, KRP-203, SEW2871, ponesimod, and MT-1303) that present leads for developing radiotracers for PET imaging of the S1P1 receptor. 188
To date, no compound has been translated for in vivo imaging of S1P1 receptors in humans, although several radiotracers have been developed and evaluated both in vitro and in vivo using murine models of MS. The S1P1 receptor antagonist [18F](R)-1-[[3-(6-fluorohexyl)-phenyl]amino-4-oxobutyl]phosphonic acid showed good in vitro potency and serum stability, but PET imaging in wild-type mice revealed increased radioactivity uptake in bones due to rapid in vivo defluorination. 189 A subsequent in vivo biodistribution study was conducted to evaluate 2 fluorinated structural analogs of FTY720 as candidate PET radiotracers. 190 Quantitative PET time–activity curves indicated a rapid clearance from blood within 3 minutes postinjection, with the declining blood concentration coincident with an intense uptake of both tracers into the liver, reflecting predominant hepatic elimination. The PET imaging results also showed positive uptake in S1P1 receptor-rich tissues, without much evidence of defluorination in vivo. However, pharmacological specificity was not formally established for these tracers.
TZ3321, a novel S1P1 receptor antagonist with high affinity in vitro (K i = 0.2 nM), was first introduced by Merck Serono. 191 The potential utility of this ligand for PET imaging was explored in a biodistribution study conducted in C57BL/6 mice, which showed excellent brain uptake of (SUV = 7.1 at 60 minutes postinjection). 192 The biodistribution results were encouraging for the use of [11C]TZ3321 as a PET tracer for S1P1 receptors. Recently, [11C]TZ3321 PET studies in the EAE rat model revealed 20% to 30% higher uptake in the lumbar spinal cord of symptomatic EAE rats versus sham controls. 193 Increased tracer uptake was correlated with increased S1P1 receptor expression in the EAE spinal cord, glial cell activation, and infiltrating IL-17–producing T cells as confirmed by immunohistochemical localization of GFAP, IBA1, and IL-17 in EAE rat spinal cord sections. Second-generation S1P1-specific 18F-labeled radiotracers were developed that demonstrated specificity for S1P1 receptors, crossed the BBB, and showed elevated binding in the murine EAE model of MS by PET. 194 In rodent biodistribution studies, the S1P1 receptor ligands [18F]TZ35110 (IC50 = 2.6 nmol/L), [18F]TZ43113 (IC50 = 9.8 nM) and [18F]TZ35104 (IC50 = 6.7 nmol/L) exhibited acceptable brain uptake with peak uptake occurring 1 hour postinjection. 194 The PET studies of [18F]TZ43113 in the female Lewis rat EAE model showed 31% higher uptake in the lumbar spinal cord. Furthermore, PET imaging of [18F]TZ35104 showed excellent uptake and washout in NHP brain. 194 The same research group then proceeded to optimize the kinetics of TZ35104, the lead structure identified from their previous study, by incorporating polyethylene glycol chains into the structures. 195 Small animal PET imaging studies with polyethylene glycol derivatives of TZ35104,such as [18F]TZ4877 and [18F]TZ4881, revealed 10% higher spinal cord uptake in the rat EAE model. 195 Further in vivo validation in different animal models of neuroinflammation as well as tracer metabolism studies are warranted for translating a suitable S1P1 receptor ligand as a PET radiotracer for clinical use in MS and other neuroinflammation conditions.
Cannabinoid-2 Receptor
The type 2 cannabinoid receptors (CB2) are G-protein (Gi/o) coupled and implicated in immunomodulation and endogenous response to injury. 196 Rat microglial cells normally express CB2 receptors, and neuroinflammatory treatments provoke upregulation of CB2 mRNA and protein, which are mainly colocalized with activated microglial cells in cerebellum, cortex, and brain stem, 197 although low levels of CB2 mRNA and protein have been detected in Purkinje cells and other neuron types of healthy brain. 198 Although the neuronal function is unclear, the CB2 receptors play an important role in microglial migration in response to inflammatory stimuli. 199 Immunohistochemical studies revealed increased CB2 receptor protein expression in microglia clustering at Aβ plaques in AD brain tissues, compared to those of healthy individuals. 200 Thus, CB2 presents an important target for PET imaging of the microglial dynamics of neuroinflammation.
Recent studies report the in vivo evaluation of potent 11C- and 18F-labeled CB2 receptor agonists. Among the candidates, [11C]NE40 demonstrated specific CB2 receptor binding in the spleen and blood of normal rats and high brain uptake in rhesus monkey. 201 Moreover, [11C]NE40 showed specific and reversible binding to CB2 receptors in a rat model with local overexpression of human CB2 receptors. [11C]NE40 uptake in the right striatum (hCB2R vector) was 2.5 times higher compared to that in the left striatum (control vector). However, results from the first-in-human PET study using [11C]NE40 failed to reveal an upregulation in patients with AD. 202 In a dual tracer study to image Aβ plaques and CB2 receptors using [11C]PIB and [11C]NE40, respectively, in patients with AD, there was no correlation between regional Aβ binding and homologous CB2 receptor availability, suggesting insufficient affinity and/or selectivity of the PET radiotracer for CB2 receptors in human brain. The same research group then developed [11C]MA2 and [18F]MA3, which are radiolabeled analogs of a highly potent N-arylamide oxadiazole CB2 agonist (EC50 = 0.02 nmol/L). 203 MA2 and MA3 display high potency for hCB2 receptors (EC50: 3 and 0.1 nmol/L, respectively), and high binding affinity in vitro for hCB2 (87 and 0.8 nmol/L, respectively). Biodistribution studies employing these radiotracers in mice demonstrated high brain uptake and efficient clearance from blood and all major organs by the hepatobiliary pathway. Taken together, [11C]MA2 and [18F]MA3 represent potential probes for PET in vivo imaging of brain CB2 receptors in neuroinflammation.
CX3CR1
CX3CR1, a Gαi-coupled 7-transmembrane chemokine receptor, is expressed in lymphocytes and microglia. 204 It has the distinction that its only known natural ligand arises from proteolysis of a transmembrane protein; the active CX3CR1 ligand, which is known as fractalkine, is shed through activity of a disintegrin/metaproteases such as ADAM17/TACE. 204 The chemokine fractalkine has the properties of a chemoattractant and adhesion molecule, being chemotactic for T cells and monocytes in inflammatory conditions such as osteoarthritis, and likewise involved in synaptic pruning and migration of microglia. 205,206 The broad-spectrum viral macrophage inflammatory protein-II linked to nanoparticles in conjunction with 64Cu has served for PET imaging of macrophages in an inflammatory atherosclerosis model. 207 Although no small molecular weight chemokine ligand has been identified for brain imaging studies, the CX3R1 antagonist AZD8797 has been characterized as a selective and orally active agent, with disease-modifying effects in a chronic-relapsing rat model for MS. 208,209 With adequate derivatization and optimization, this work might serve to identify lead compounds for development of a brain-penetrating PET tracer for the CX3R1.
Purinergic Receptors—P2X7 and P2Y12
Purinergic signaling is mediated by the P2 family of receptors that plays a crucial role in physiology of the nervous system. 210 P2 receptors are broadly categorized into 2 types: (1) P2X ionotropic receptors, which are ATP-gated ion channels and (2) P2Y metabotropic receptors, which are G-protein–coupled receptors such as the inhibitory adenosine receptors blocked by caffeine.
The P2X7 receptor, a subtype of P2X ionotropic receptors, comprises 2 transmembrane domains, an extracellular loop (with the ATP-binding site), and intracellular N- and C-terminal domains. 211 In brain, the P2X7 is predominantly expressed in activated microglia and to a lesser extent in neurons and astrocytes, being most abundant in spinal cord, cerebellum, hypothalamus, and substantia nigra. 212 -214 Under normal physiological conditions, P2X7 is considered a “silent receptor,” but it is functionally upregulated during pathological states. Following an insult, ATP is released into the extracellular space at higher concentrations, where it initiates inflammatory signaling cascades, principally via P2X7. 215 Overexpression of P2X7 receptors in rat primary hippocampal cultures was shown to drive the activation and proliferation of microglia in the absence of pathological insults. 216 Persistent elevations in extracellular ATP and proinflammatory cytokines will promote neuroinflammation and neurodegeneration. The expression and functioning of P2X7 receptors is significantly upregulated in postmortem brain of patients with AD as well as various neurodegenerative disease animal models. 217 Given this background, pharmacological suppression of the P2X7 pathway may offer a novel therapeutic approach for ameliorating neuroinflammatory processes.
A-740003, a selective P2X7 receptor antagonist (IC50: rP2X7R = 18 nmol/L; hP2X7R = 40 nmol/L) was pursued in the first efforts to develop P2X7 receptor imaging by PET. 211 Biodistribution studies in healthy rats revealed only traces of [11C]A-740003 uptake, either due to poor BBB permeability or due to low receptor expression levels in healthy brain. The same research group then developed an P2X7 receptor allosteric antagonist, [11C]SMW139. 218 The PET imaging studies conducted with [11C]SMW139 in rats overexpressing the hP2X7 receptor in the right striatum demonstrated 1.5-fold higher radioactivity uptake in the right striatum compared to the contralateral striatum, and this effect was blocked by pretreatment with JNJ-4796556, a potent P2X7 receptor antagonist.
JNJ-54173717, which has nanomolar affinity for rat P2X7 R and hP2X7 R, 219,220 was radiolabeled with 11C and evaluated in vivo in rats and NHPs. 221 Biodistribution studies conducted in normal rats demonstrated that [11C]JNJ-54173717 readily crossed the BBB and was cleared from plasma mainly via the hepatobiliary pathway. 221 To conduct further in vivo evaluation and validation of this PET tracer, a humanized animal model was developed in which hP2X7 R was overexpressed in the rat striatum after stereotaxic injection of viral vectors. 221 The PET studies in these rats showed binding of [11C]JNJ-54173717 in the striatum expressing hP2X7 R, with rapid washout from the noninjected control striatum and other brain regions. Furthermore, in the NHP brain, pretreatment with cold JNJ-54173717 and with the P2X7 R antagonist JNJ-42253432 reduced uptake of [11C]JNJ-54173717, suggesting P2X7R-selective binding of this radiotracer. 221 This radioligand appears to be a promising candidate for further evaluation of P2X7 R expression levels in neurodegenerative disorders.
The potent P2X7 antagonist GSK1482160 (IC50hP2X7R = 3 nM) was labeled with carbon-11 to investigate its fitness as a radiotracer for PET imaging. 222 Biodistribution studies with [11C]GSK1482160 revealed increased radiotracer accumulation in brain of LPS-treated mice compared to control animals 223 ; this effect was blocked by pretreatment with an excess of authentic standard GSK1482160. The PET studies demonstrated slow in vivo kinetics and homogeneous distribution in the brain of healthy NHP, but responses to a neuroinflammatory challenge were not investigated. 224 In the EAE rat model, uptake of [11C]GSK1482160 in lumbar spinal cord was the highest at the EAE-peak symptomatic stage, when radiotracer uptake correlated strongly with P2X7-positive cell counts, activated microglia numbers, and disease severity.
The P2Y12 receptor is an adenosine diphosphate-responsive G-protein–coupled receptor that is selectively overexpressed on activated microglia of the anti-inflammatory M2 phenotype. 225,226 There is sparse P2Y12 mRNA expression throughout the normal adult rat brain, including the neocortex, hippocampus, cerebellum, and brain stem. 227 Immunohistochemical studies showed decreased P2Y12 receptor expression in microglia near the Aβ plaques in postmortem brains from patients with AD and near the demyelinated lesions in patients with MS. 225 This decrease in P2Y12 receptor expression in such neuroinflammatory diseases implies a shift in the balance away from anti-inflammatory to proinflammatory M1 microglial cells near the lesions.
[11C]
Receptor for Advanced Glycation End Products
The RAGE is a 45- to 55-kDa member of the immunoglobulin receptor superfamily with 3 extracellular domains, a transmembrane domain, and a highly charged intracellular domain that mediates interaction with oxidative stress-related signal transduction molecules (eg, mitogen-activated protein kinase and nuclear factor-κB). 229,230 In addition to advanced glycation end products, RAGE binds several other ligands, including Aβ, a central player in AD pathogenic mechanisms. 231 -234 Two functional types of RAGE have been associated with neurological diseases: cell membrane-bound full length (flRAGE) and soluble (sRAGE). 235 In general, endogenous ligand binding to flRAGE initiates receptor-dependent signaling, resulting in inflammation and cellular stress. This process engages a positive feedback mechanism that can accelerate disease progression. In contrast, sRAGE lacks the cellular signaling domain but usually retains the ligand-binding domain whereby they can sequester endogenous ligand, thus preventing it from binding to flRAGE and activating damaging proinflammatory pathways. 235 Clearly, sRAGE constitutes a naturally occurring “governer” that prevents uncontrolled RAGE-mediated inflammation and cellular stress. The RAGE is constitutively expressed on microglia and neurons of the hippocampus, entorhinal cortex, and superior frontal gyrus. 236 Under normal physiological conditions, flRAGE is expressed at low levels, but its expression appears to increase with aging and in AD. 235 -237 On the other hand, levels of sRAGE serve as a predictive marker for disease protection; compared to cognitively normal controls, sRAGE levels were lower in circulation of patients with AD and in individuals with early cognitive decline. 238,239 Circulating sRAGE is derived from enzymatic cleavage of flRAGE from the plasma membrane. 235 A decline in circulating sRAGE levels along with progressive cognitive decline in AD suggests that an imbalance between circulating sRAGE and endogenous RAGE ligands may influence or exacerbate pathological events in the brain. Whether monitoring the changes in sRAGE levels in the circulation can serve as an indicator for disease development and severity requires further investigation. Nevertheless, centrally acting RAGE antagonists or sRAGE mimetics may serve as novel therapeutic approaches for neuroinflammation.
[18F]RAGER exhibited high affinity (K d = 15 nmol/L) for RAGE in vitro in both AD and control tissues and was the first small-molecule radiotracer that was explored for in vivo PET imaging. 240 The PET studies conducted in rodents and NHP confirmed BBB permeability and increased uptake in the brain areas known to express RAGE. Autoradiographic images of [18F]RAGER binding in postmortem brain revealed increased specific biding in frontal cortex gray matter from patients with AD compared to normal control donors. Moreover, [18F]RAGER exhibited an in vitro B max/K D ratio of 2 in AD tissue, predicting a sufficient BP in vivo to justify using this scaffold for future applications in preclinical and clinical PET imaging. 240 As a step toward translation of [18F]RAGER into clinical PET studies, a CNS receptor screening was performed to identify any off-target binding. 241 Results of this screening showed that RAGER exhibited 93 nmol/L affinity toward the human melatonin MT1 receptor (in vitro). Nevertheless, competitive blocking studies with melatonin revealed that [18F]RAGER binding was specific to RAGE, as melatonin did not displace [18F]RAGER binding in human brain tissue autoradiography studies. Moreover, pretreatment with melatonin (10 μg/kg) failed to displace [18F]RAGER binding in both rodent and NHP PET blocking studies. Further investigations are needed to confirm the selectivity of this tracer for either flRAGE or sRAGE subtypes, as this will enable monitoring alterations in the 2 RAGE subtypes in relation to disease progression and severity.
In order to explore the potential of RAGE as a new PET biomarker of AD diagnosis and monitoring, carbon-11-labeled RAGE inhibitor, [11C]FPS-ZM1, was evaluated in autoradiography studies using brains from wild-type and AD mice. 242 Results from these studies highlighted high nonspecific binding in both wild-type and AD mice, thereby warranting further structural optimization. [18F]FPS-ZM1 was developed to explore the link between diabetes and AD. 243 Biodistribution of [18F]FPS-ZM1 in healthy C57BL/6J mice displayed high brain permeability followed by rapid clearance from brains and most other organs (except for liver, cecum, and gonad), indicating low nonspecific binding in brain and majority of the peripheral organs. Small animal PET imaging indicated that the uptake of [18F]FPS-ZM1 in brain was higher in dementia models than in type 1 and type 2 diabetic models, suggesting that [18F]FPS-ZM1 might be a promising biomarker for early detection of AD in patients with diabetes. Overall, a number of chemical entities that block RAGE activation by binding to the extracellular or intracellular domains have been reviewed elsewhere, 244 which may act as scaffolds for further chemical refinement and optimization of PET radiotracers that would elucidate alterations in RAGE expression and distribution in both healthy and pathological conditions.
Mer Tyrosine Kinase
The MerTK is a cell surface receptor tyrosine kinase that plays a critical role in the recognition, phagocytosis, and clearance of apoptotic cells in a variety of tissues. 245,246 In brain, MerTK is predominantly expressed in the microglia and astrocytes and is implicated in neuroinflammation and oncogenesis. 246,247 While MerTK protein levels are low to undetectable in normal brain, increased expression has been demonstrated in various neuroinflammatory disorders. For example, high levels of the soluble form of MerTK were found in a postmortem analysis of established MS lesions. 248 Also, polymorphisms in the MerTK gene are reportedly associated with MS susceptibility. 249 Although the precise role of MerTK in neuroinflammation remains unclear, PET imaging of this target should assist in devising new targeted therapeutic strategies for neuroinflammation and associated diseases.
[18F]JHU16907, the first tracer developed for PET imaging of MerTK, was derived from a potent MerTK inhibitor 2-fluropyridine derivative, JHU16907 (IC50 = 2.5 nmol/L). 250 Robust brain uptake of [18F]JHU16907 was found in control CD-1 mice, with peak at 5 minutes postinjection (SUVpeak 3% ID/g) cerebellum, hippocampus, and throughout cortex, followed by rapid washout. However, regional distribution of radioactivity showed relatively low heterogeneity, consistent with the globally low MerTK expression in healthy brain. 250 [18F]JHU16907 was further evaluated in a biodistribution study conducted in an LPS rodent model of neuroinflammation. This study showed higher cerebral uptake of [18F]JHU16907 in LPS-treated CD-1 mice than in control CD-1. Moreover, self-blocking studies (1 or 3 mg/kg JHU16907) significantly displaced [18F]JHU16907 binding of LPS-treated mouse, confirming the specificity of this tracer for MerTK in vivo. There is considerable scope for imaging MerTK, thus warranting an extended search for improved MerTK tracers to enable eventual translational imaging research in human disease.
Triggering Receptor Expressed on Myeloid Cells-1
The TREM family of proteins comprises a group of cell surface innate immune receptors that are expressed on various myeloid cell populations throughout the body, including microglia in the brain. 251,252 The 2 subtypes of TREM, namely, TREM1 and TREM2, have distinct roles in immune function. Normal brain expresses very low to undetectable levels of TREM1. However, mice brain cell suspensions obtained 24 hours after intracerebral LPS injection revealed a significant increase in TREM1 expression and suppression in TREM2 expression as evidenced by gene expression analysis using qualitative polymerase chain reaction. 252 The differential expression of TREM1 and TREM2 in response to an acute inflammatory insult highlights the necessity to further understand how TREM expression is regulated in neurodegenerative diseases.
Initial efforts have been made toward developing brain-penetrating radiotracers for PET imaging of TREM1. A TREM1-specific PET tracer was recently developed by radiolabeling a selective anti-TREM1 antibody with copper-64 (64Cu, t ½ = 12.7 hours). 253 Specificity of this PET radiotracer was verified in vitro by HEK293 cells with and without TREM1 transfection. Furthermore, in vivo PET imaging studies were conducted in different murine models of neuroinflammation, including LPS-induced systemic inflammation and ischemic stroke. 253,254 This thorough in vivo evaluation demonstrated higher binding of [64Cu]TREM1-mAb in brain regions exhibiting inflammation in all mouse models investigated, compared to control mice. In vivo specificity of the PET radiotracer was further confirmed by studies employing TREM1 knockout mice. These results seem remarkable, given the caveat that the BBB is not usually permeable to large molecular weight tracers. Nonetheless, TREM1 knockout mice treated with LPS or following EAE induction exhibited negligible cerebral binding of [64Cu]TREM1-mAb, thereby highlighting the specificity of the PET radiotracer. Overall, these results present a promising start for in vivo imaging of TREM1 and investigating its role in neuroinflammation, although there likely remains a need for lower molecular weight ligands to achieve high specific binding of the PET ligand in vivo.
Conclusion
Neuroinflammation is a complex, orchestrated cellular response to harmful stimuli, which under acute conditions is aimed toward restoring tissue homeostasis. However, when inflammation is unresolved and becomes chronic, the otherwise salutogenic responses increase tissue damage and promote neurodegenerative disease initiation/progression. Emerging evidence suggests that chronic activation of microglia and astrocytes plays a more central role in neurodegenerative diseases than previously thought. Despite decades of research and the pharmaceutical industry’s persistent efforts toward the discovery and development of treatment options for neurodegenerative diseases, translation of therapies to the clinic has faced considerable failure. 255 Taking as an example the case of AD, over 200 compounds have reached phase II clinical trials for AD since 2003, but no new drugs have been approved. 256,257 Nevertheless, important lessons have been learned from failures of anti-AD therapies in late-stage clinical trials, including dimebon (Medivation and Pfizer) and solanezumab (Eli Lilly). Therapy applied in late stages of AD seems unlikely to be ineffective, since ongoing chronic neuroinflammatory processes will have caused irreversible damage. Patients are generally recruited to trials only after their plaque burden and neurodegeneration has advanced and the disease is probably irreversible. 258 Moreover, detailed stratified analysis of clinical data reveals more promising results for treatment of patients with AD in earlier stages of the disease. 259 High heterogeneity in microglia and astrocyte reactions, as evidenced by molecular profiling in mouse models of neurodegenerative diseases, suggests that failure of neuroinflammation-targeted therapies might be due to their lack of immunoselectivity and inability to restore CNS cell function. 255 Clearly, the clinical development plan for neurodegenerative diseases is being revisited, and potential early diagnostic biomarkers and preventative strategies are being explored. To address the failures associated with AD drug development, the Committee for Medicinal Products for Human Use proposed changes in the diagnostic criteria, and more importantly encouraged the use of biomarkers in different stages of drug development. 260 In vivo imaging of biomarkers of neuroinflammation should assist in early detection of AD and other neurodegenerative diseases and help clinical researchers measure early responses to therapeutic interventions. 261 It is also noteworthy that neurodegenerative diseases involve multiple pathologies; therefore, a combination therapy appears to be a rational approach for treatment. 262 Given the significant role of neuroinflammation in the pathogenesis of neurodegenerative diseases, PET imaging biomarkers of neuroinflammation will not only assist in tracking disease progression but will also guide the design of combination therapies in an efficient and cost-effective way.
For 2 decades, TSPO PET imaging of neuroinflammation has held center stage in studies of neuroinflammatory diseases, despite several caveats related to quantitation, allelic dependence of binding, microglial phenotype, and incomplete cellular specificity for microglia. Nonetheless, TPSO PET imaging has, for example, highlighted the relationship between Aβ accumulation and microglial activation in AD, 263 and PET imaging of microglial activation, beyond TSPO, has recently been reviewed for CB2, COX-2, the P2X7 receptor, and ROS. 264 Other PET targets discussed herein, such as GSK-3, MAO-B, COX, and S1P1, remain in a relatively preliminary stage of development and implementation as neuroimaging targets, but preclinical findings suggest that they offer immense potential for PET radiotracer development and clinical translation. In addition to the emerging neuroimmune targets discussed herein, ion channel linked receptors that include γ-aminobutyric acid—benzodiazepine receptor and nicotinic acetylcholine receptors (α7 and α4β2 subtypes)—also present promising potential for PET imaging, although further preclinical and clinical research is needed to establish the utility of these radiotracers for probing neuroinflammation and associated neurological disorders. We and others have previously reported selected PET radioligands developed for these targets. 265 -268 Development of receptor subtype-specific radiotracers while maintaining or improving brain uptake remains a major challenge for PET radioligand development for these targets. Nevertheless, recent human PET imaging studies measuring α7 269 and α4β2 270 nicotinic acetylcholine receptor distribution in healthy volunteers using newly developed radioligands with superior in vivo characteristics support the translational potential of these targets for PET imaging of neurodegenerative diseases.
Alternate approaches are concurrently under development to identify clinically useful inflammatory cytokines in the plasma and establish the relationship between their peripheral and central distribution. 271 In this approach, proinflammatory biomarkers such as IL-1β, interferon-γ, IL-10, and IL-6 can be assessed in plasma and cerebrospinal fluid samples using Multiplex cytokine and proinflammatory biomarker analyses. Combining PET imaging results with proinflammatory biomarker analyses of plasma and cerebrospinal fluid samples from the same individuals in vivo may elucidate the molecular inflammatory signature in early-stage neurodegenerative disease and may assist in efficient translational application of relevant therapeutic interventions. 271 We contend that PET imaging of multiple biomarkers across the course of neurodegenerative disease will contribute toward better diagnosis and tracking of the sequence of pathophysiological events. One major remaining hurdle entails the distinction between the M1 and M2 microglial phenotypes. The P2Y12 purinergic receptor is selectively expressed on M2 (neuroprotective) microglia, and PET imaging of this target may be especially beneficial in evaluating efficacy of potential therapies. For example, an anti-inflammatory therapy should ideally upregulate P2Y12 expression, thus engaging a potentially neuroprotective mechanism. As discussed earlier, radiotracer development for PET brain imaging is a very challenging task. A fundamental criterion for a candidate radioligand is its ability to cross the BBB. Some of the lead structures highlighted in this review displayed favorable in vitro binding properties, including good binding potential and selectivity. Nevertheless, upon transition to in vivo PET imaging, the radiotracer failed to display optimum BBB penetrability. Future studies are needed to acquire sufficient information to predict a radiotracer’s brain penetrability, efflux protein interaction, and metabolism and to find a radiotracer with adequate specific binding and ideal metabolism that can overcome hurdles for human translation.
In conclusion, PET radiotracer development for neuroinflammation beyond TSPO, for imaging neurodegenerative diseases (and other neurological disorders), represents an active area of research with several emerging targets and tracers under various stages of preclinical and clinical research studies underway. Continuing development of PET radiotracers for imaging of neuroinflammation presents an attractive and noninvasive approach that can enable early diagnosis, track disease progression, and aid in the rational design and clinical assessment of patient responses to therapeutic intervention.
Footnotes
Acknowledgments
NV thanks National Institute on Ageing of the NIH (R01AG054473), the Azrieli Foundation, and the Canada Research Chairs Program for support.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.