
Abstract
Global cerebral ischemia induced by cardiac arrest usually leads to poor neurological outcomes. Numerous studies have focused on ways to prevent ischemic damage in the brain, however clinical therapies are still limited. Our previous studies revealed that delta opioid receptor (DOR) activation with [d-Ala2, d-Leu5] enkephalin (DADLE), a DOR agonist, not only significantly promotes neuronal survival on day 3, but also improves spatial memory deficits on days 5-9 after ischemia. However, the neurological mechanism underlying DADLE-induced cognitive recovery remains unclear. This study first examined the changes in neuronal survival in the CA1 region at the advanced time point (day 7) after ischemia/reperfusion (I/R) injury and found a significant amelioration of damaged CA1 neurons in the rats treated with DADLE (2.5 nmol) when administered at the onset of reperfusion. The structure and function of CA1 neurons on days 3 and 7 post-ischemia showed significant improvements in both the density of the injured dendritic spines and the basic transmission of the impaired CA3-CA1 synapses following DADLE treatment. The molecular changes involved in DADLE-mediated synaptic modulation on days 3 and 7 post-ischemia implied the time-related differential regulation of PKCα-MARCKS on the dendritic spine structure and of BDNF- ERK1/2-synapsin I on synaptic function, in response to ischemic/reperfusion injury as well as to DADLE treatment. Importantly, all the beneficial effects of DADLE on ischemia-induced cellular, synaptic, and molecular deficits were eliminated by the DOR inhibitor naltrindole (2.5 nmol). Taken together, this study suggested that DOR activation-induced protective signaling pathways of PKCα-MARCKS involved in the synaptic morphology and BDNF-ERK-synapsin I in synaptic transmission may be engaged in the cognitive recovery in rats suffering from advanced cerebral ischemia.
Introduction
Cardiac arrest (CA) is one of the leading causes of morbidity and mortality worldwide. In China, there are more than 230 million people with cardiovascular disease, and 550,000 individuals experience CA every year 1 . Although the overall incidence of return of spontaneous circulation (ROSC) among CA patients significantly improved over the 40-year period 2 , many patients remain persistently comatose or have a poor neurological outcome due to cerebral hypoxia 3 . Numerous studies have focused on ways to prevent ischemic damage in the brain, yet clinical therapies remain limited. Thus, there is a critical need for basic investigations into the pathological processes, molecular mechanisms, and potential therapeutic strategies for ischemic disease.
The delta opioid receptor (DOR) is a G protein coupled receptor that belongs to the opioid system. DOR is broadly expressed in the brain, especially in the hippocampus, which is known to contribute to learning and memory. Growing evidence has revealed the neuroprotection of DOR in pathological pain, anxiolytic effects, addiction, and Parkinson’s disease 4 –7 . In recent years, interest has grown in DOR-mediated neuroprotection against hypoxic/ischemic injury 8 . For instance, DOR activation may reduce ischemic volume and attenuate neurological deficits in a focal ischemic model using the non-peptidic DOR agonist Tan-67 9,10 . Similar neuroprotective effects have also been observed in global ischemic animals by hypoxic preconditioning (HPC) 11 . Studies have shown that the neuroprotection from DOR activation is related to the modulation of several signaling pathways, such as brain-derived neurotrophic factor (BDNF)-tyrosine kinase receptor B (TrkB) 12 , protein kinase C (PKC)/extracellular regulated protein kinases (ERK), 13 and phosphoinositide 3 kinase (PI3 K)/protein kinase B (Akt) 14,15 . These results suggest that DOR may be a potential target for cerebral ischemia and cognitive impairment.
Evidence suggests that cerebral ischemia may be detrimental to the structural stability of synapses, which may subsequently impair synaptic transmission 16 . Synaptic activity can dynamically modulate protein synthesis and reorganization of cytoskeletal architecture in dendritic spines, which affects both the plasticity of the structure and the function of the spines 17 . Several important molecules have been shown to be involved in the modulation of synaptic plasticity, such as BDNF, an important regulator of synaptic transmission and long-term potentiation (LTP) in the hippocampus, which is coupled with the activation of the Ras/ERK, PI3K/Akt, and phospholipase C-γ (PLC-γ) pathways 18 . In addition, the PKC family plays a vital role in modulating neuronal membrane structural events. For example, PKCα activation causes neurite outgrowth and retraction in rat hippocampal neurons 19 and other cell types 20 .
Our previous studies demonstrated that DOR activation with [d-Ala2, d-Leu5] enkephalin (DADLE) improved cognition impairment, reduced neuronal death, and regulated the activation of astrocytes in global ischemic rats 21,22 . However, the structural and functional synaptic changes in response to ischemia/reperfusion (I/R) injury and DADLE treatment in synaptic changes remain unexplored and the relevant molecular mechanisms remain unclear. In this study, we investigated the neuroprotective effects of DOR against I/R injury by focusing on the PKC-mediated neuronal morphological recovery and BDNF-mediated synaptic transmission improvement in ischemic rats following DADLE treatment.
Materials and Methods
Animals
All animal experiments were performed in accordance with the Animals Act (2006, China) and approved by the Institutional Animal Care and Use Committee (IACUC approval ID #AR201404022) of East China Normal University. Male Sprague-Dawley rats (mean body weight 280-320 g) were purchased from the Shanghai Laboratory Animal Center (Shanghai, China). The rats were individually housed and maintained on a 12 h light/dark cycle under constant temperature and humidity. The animals had free access to water and food. Efforts were made to minimize the number of animals used and their suffering.
Global Cerebral Ischemia Model
Global ischemia was induced in rats by four-vessel occlusion (4-VO), as described previously 21 –23 . Briefly, the rats were anesthetized with 10% chloral hydrate (300 mg/kg, i.p.), and the bilateral vertebral arteries were electrocauterized with an electrocoagulator through the alar foramina of the first cervical vertebra. Both common carotid arteries were exposed, and a small-diameter silk thread was looped around each carotid without interrupting blood flow to facilitate subsequent occlusion. The animals were anesthetized and fasted prior to the next operation. The next day, the rats were lightly anesthetized with ethyl ether, and the bilateral common carotid arteries were re-exposed. After the rats were awake, the bilateral common carotid arteries were occluded for 10 min with artery clamps. Reperfusion was induced by releasing artery clamps after 10 min of ischemia. Ischemia was ensured by monitoring the loss of the righting reflex and bilateral pupil dilation during carotid occlusion. Sham-operated rats underwent the same surgical procedures, except for carotid artery occlusion. Core body temperatures were monitored with a rectal probe and maintained at 37°C throughout the experiment using a heating lamp.
Intracerebral Cannula and Groups
Rats were anesthetized with 10% chloral hydrate and implanted with a cannula into the right lateral ventricle (Fig. 1), and placed in a stereotaxic frame with a horizontal skull position. A guide cannula was positioned using the following coordinates: 0.8 mm posterior to bregma, 1.5 mm lateral to the midline, and 3.8 mm ventral to the skull surface. All coordinates were derived from the atlas of Paxinos and Watson 24 . The guide cannula and two stainless steel screws were anchored to the skull using acrylic dental cement. Following at least 6 days of recovery, the rats underwent 4-VO surgery, as described above.
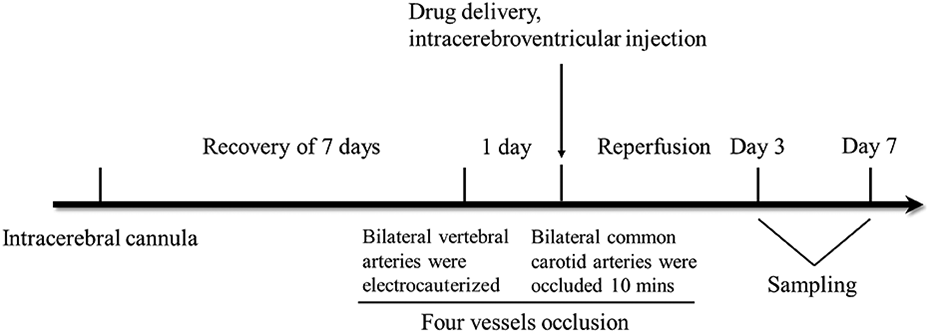
The diagram of experimental procedure, including intracerebral cannula, global ischemia, drug delivery and sampling.
A total of 120 rats were divided into four groups (n = 30 per group): the sham group (sham procedure with intracerebroventricular injection of PBS), I/R group (ischemia procedure with intracerebroventricular injection of PBS), DADLE-treated group (ischemia procedure with intracerebroventricular injection of DADLE), and DADLE+Naltrindole group (ischemia procedure with intracerebroventricular injection of DADLE and naltrindole). For drug delivery in DADLE postconditioning, a total volume of 5 µl PBS or PBS containing 2.5 nmol DADLE (Sigma, USA) or PBS containing 2.5 nmol DADLE and 2.5 nmol Naltrindole (Sigma, USA) was administered slowly at the onset of reperfusion.
Perfusion and Tissue Preparation
Seven days after ischemia, the rats were anesthetized with 10% chloral hydrate and perfused transcardially with normal saline followed by 4% ice-cold paraformaldehyde (PFA). The brains were then removed and immediately submerged in 4% PFA solution at 4°C overnight. The brains were dehydrated using a sucrose gradient. The 30 µm coronal sections of the brain containing the hippocampus were prepared and stored at −80°C.
Nissl Staining
Nissl staining was used to detect Nissl bodies in the cytoplasm of neurons. Frozen sections were mounted on gelatin-coated slides, dried at room temperature, and then placed in distilled water for 2 min. After staining in warmed 0.5% cresyl violet solution for 10 min, the slides were quickly rinsed with distilled water and differentiated twice in 95% ethyl alcohol for 2 min each. The slides were cleared twice in xylene, each for 5 min, mounted with neutral balsam, and then examined under a microscope. Hippocampal neurons were counted in the middle of the linear part of eight different CA1 fields from each animal and expressed per 1 mm of the hippocampal CA1 sector. A positive cell count was performed by an investigator who was blinded to the treatment groups using image tool software (Nikon NIS-Elements, USA).
Golgi Staining
Golgi impregnation method was used to visualize the dendritic spines of rat hippocampal neurons, according to the manufacturer’s instructions (FD Rapid Golgi Stain Kit, USA). Golgi staining tissues were analyzed using a light microscope (Leica, Germany). Whole hippocampus images were acquired in a 100× magnification lens. Dendritic spines in CA1 neurons were observed in the 1000× oil immersion lens. Dendritic spine density was analyzed by the number of spines on a limited length of a dendrite by two independent investigators who were blinded to the treatment groups. At least two dendritic segments of at least 10 µm were analyzed per CA1 neuron, and five neurons in the CA1 region of individual rats (n = 5 rats per group) were examined for dendritic spine density.
Brain Slice Preparation
Rats were anesthetized with 10% chloral hydrate and killed by decapitation. The brain was cut into coronal slices (370 mm thickness) containing the hippocampus with ice-cold (4°C) and oxygenated (95% O2 /5% CO2) modified ACSF (Choline chloride 110 mM, KCl 2.5 mM, CaCl2 0.5 mM, MgSO4 7 mM, NaHCO3 25 mM, NaH2PO4 1.25 mM, D-glucose 25 mM; pH 7.4). The slices were placed in a holding chamber containing oxygenated ACSF (NaCl 119 mM, KCl 2.5 mM, CaCl2 2.5 mM, MgSO4 1.3 mM, NaHCO3 26.2 mM, NaH2PO4 1 mM, D-glucose 11 mM; pH 7.4) and left to recover for 1 h at 31°C before recording.
Field Potential Recording
For field potential recording, slices were transferred to a recording chamber with oxygenated ACSF, and the rate of ACSF superfusion was 0.5 mL/min. A unipolar stimulating electrode was placed in the stratum radiatum to activate the Schaffer collateral pathway projecting to the CA1. The field excitatory post-synaptic potentials (fEPSPs) were recorded using a glass microelectrode filled with 0.1 M CH3COONa (3-5 MΩ). The range of the input/output curve was from 0.1 mA to 1.0 mA. Paired-pulse response (PPR) was induced by paired-pulse simulation with interpulse intervals (IPIs) of 20, 40, 60, 80, 100, 200, 350, 400, 600, 800, and 1000 ms. Paired-pulse facilitation (PPF) was evaluated by the ratio of the amplitude of the second fEPSP to that of the first fEPSP elicited by pairs of stimuli (pulse 2/pulse 1 × 100). Synaptic responses were monitored with stimuli consisting of constant current pulses (0.05 ms, 0.033 Hz).
Immunoblotting
The hippocampus was rapidly isolated and protein concentrations were determined using a BCA detection kit. Equal amounts of protein were separated by 10–12% SDS-PAGE and electrotransferred onto a PVDF membrane. After blocking with 5% skim milk, the membranes were incubated with primary antibodies against BDNF (1:100, Santa Cruz Biotechnology), p-ERK1/2, PKCα, β-actin, GAPDH (1:1000, Cell Signaling Technology), p-synapsin I, phospho-myristoylated alanine-rich C kinase substrate (p-MARCKS, 1:1000, Signalway Antibody), PKCε, PKCγ (1:1000, Proteintech Group) overnight at 4°. Following incubation with the secondary antibodies for 2 h at room temperature, the membranes were scanned using a Bio-Rad gel imaging system.
Statistical Analysis
All analyses were performed using GraphPad Prism 7 (San Diego, CA, USA). Values are expressed as the mean ± SEM. Multiple data sets were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test. A two-way repeated measures ANOVA was used for the input-output curve and paired-pulse facilitation analysis. Differences were considered significant at P < 0.05 (indicated by *).
Results
Survival of Hippocampal Neurons
As an extension of our previously work that showed DADLE treatment could significantly promote neuronal survival following 3 days of I/R injury, we assessed the neuronal protection of the DOR agonist in the hippocampus of rats after 7 days of I/R treatment. The results showed distinct neuronal death in the CA1 region in the I/R group compared with the sham group. DADLE treatment significantly rescued the neuronal loss compared to the I/R group, while the protective roles were abolished by the addition of naltrindole, a DOR inhibitor, as shown in the DAD+Nal group (Fig. 2).
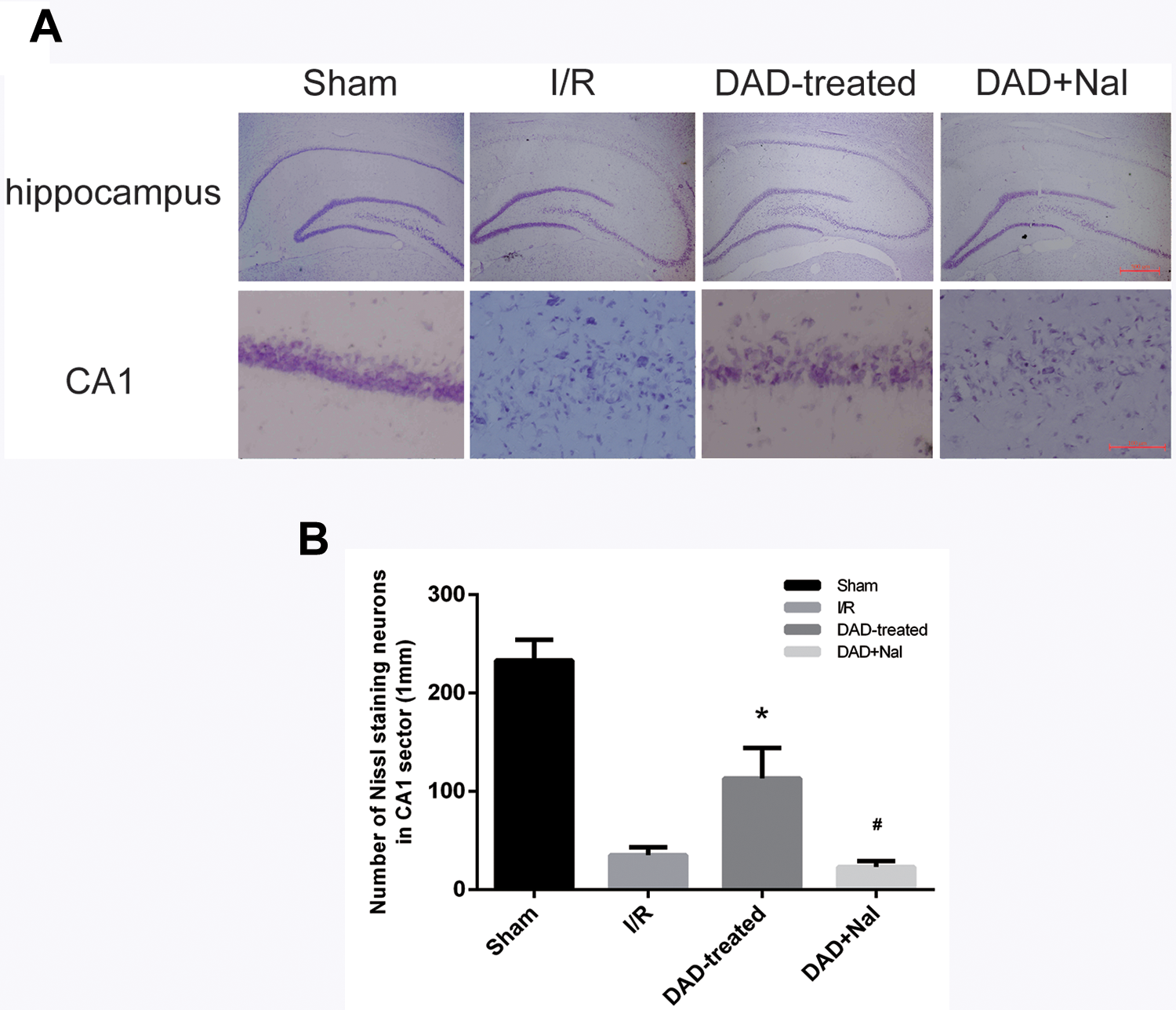
The effects of DADLE treatment in hippocampal neurons on day 7 after global ischemia. The represent images of hippocampal neurons in sham, I/R, DAD-treated and DAD+Nal group. In the CA1 region, marked neuronal loss was observed in the I/R group and the DAD+Nal group compared with the sham group. Neuronal survival was significantly increased in the DAD-treated group compared to I/R group. (Upper panel: 40× magnification; Lower panel: 400× magnification; N = 5 per group)
Morphological Plasticity of Hippocampal Neurons
To better explain the protective effects of DADLE on ischemic neurons, the changes in morphology of the CA1 neurons on days 3 and 7 post-ischemia were further examined. As indicated by Golgi staining assays, the dendritic spines of CA1 neurons markedly decreased in the I/R group compared to the sham group, and significantly increased by DADLE treatment in the rats on day 3 (Fig. 3) and day 7 (Fig. 4) after ischemia. In contrast, the DADLE-induced morphology recovery was faded in the DAD+Nal group (Fig. 3, 4).
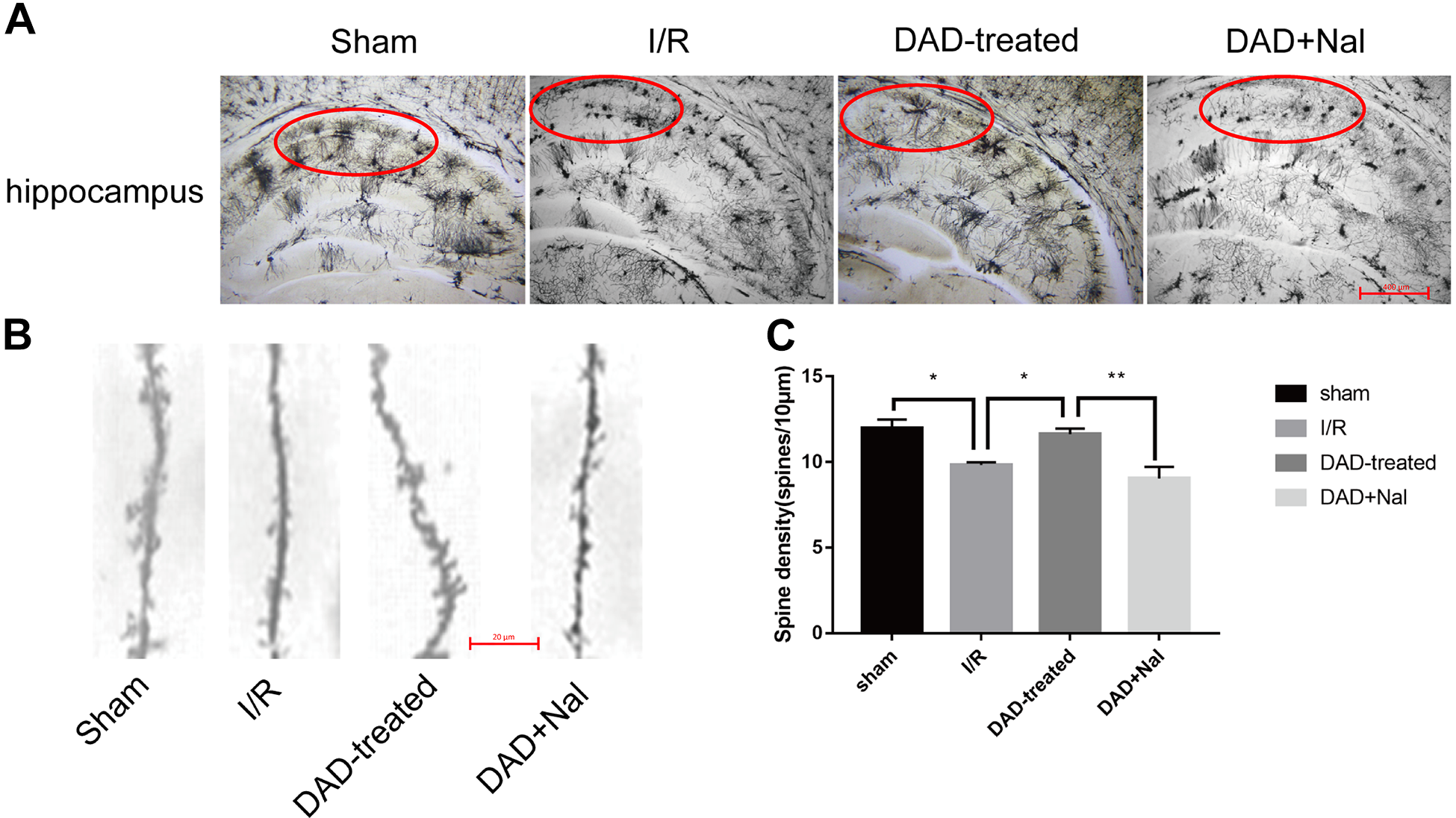
The morphological changes of dendritic spines in hippocampal neurons on day 3 after global ischemia. (A) The Golgi stain results of hippocampal neurons on day 3 after global ischemia. Red circles represent CA1 region. (100× magnification; N = 5 per group). (B) The represent images of dendritic spines in CA1 neurons on day 3 after global ischemia (1000× magnification). (C) The quantitative analysis of spine density in CA1 neurons on day 3 after global ischemia. (N = 5 per group). *P < 0.05. **P < 0.01.
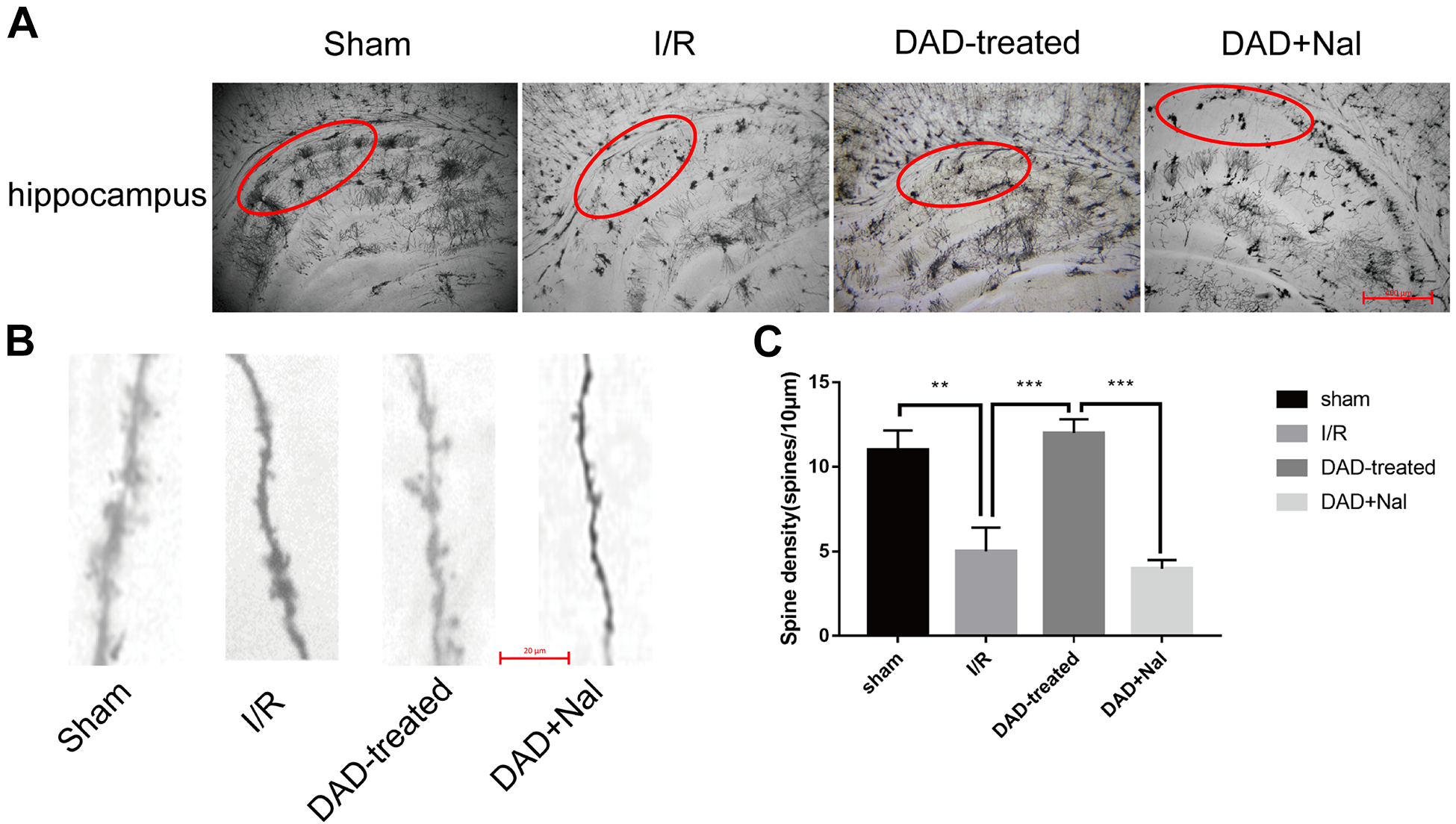
The morphological changes of dendritic spines in hippocampal neurons on day 7 after global ischemia. (A) The Golgi stain results of hippocampal neurons on day 7 after global ischemia. Red circles represent CA1 region. (100× magnification; N = 5 per group). (B) The represent images of dendritic spines in CA1 neurons on day 7 after global ischemia (1000× magnification). (C) The quantitative analysis of spine density in CA1 neurons on day 7 after global ischemia. (N = 5 per group). *P < 0.05. **P < 0.01. ***P < 0.001.
Transmission Plasticity of Hippocampal Neurons
We previously found that DADLE can rescue the cognitive impairments of rats after 5-9 days of I/R injury 21 , and given the fact that the delta opioid peptide can significantly improve neuronal morphological plasticity in the rats, particularly on day 7 after ischemia as described above, we continued to find out if there might be a connection of these results with the changes in synaptic plasticity of the hippocampal neurons at a matched time point, that is, on day 7 post-ischemia. Basal synaptic transmission in the CA3-CA1 synapses of the hippocampus was measured by in vitro electrophysiological recording. Significant differences were observed in the input-output curves (Fig. 5A) and PPR (Fig. 5B) between I/R and DAD-treated groups of rats, and the beneficial effects of DADLE on the transmission plasticity were eliminated by the addition of Naltrindole, as evident in the DAD+Nal group (Fig. 5). These results suggest that, in addition to its role in morphological plasticity, DADLE-induced DOR activation affects basal synaptic transmission, thereby contributing to cognitive recovery in rats with a prolonged I/R injury.
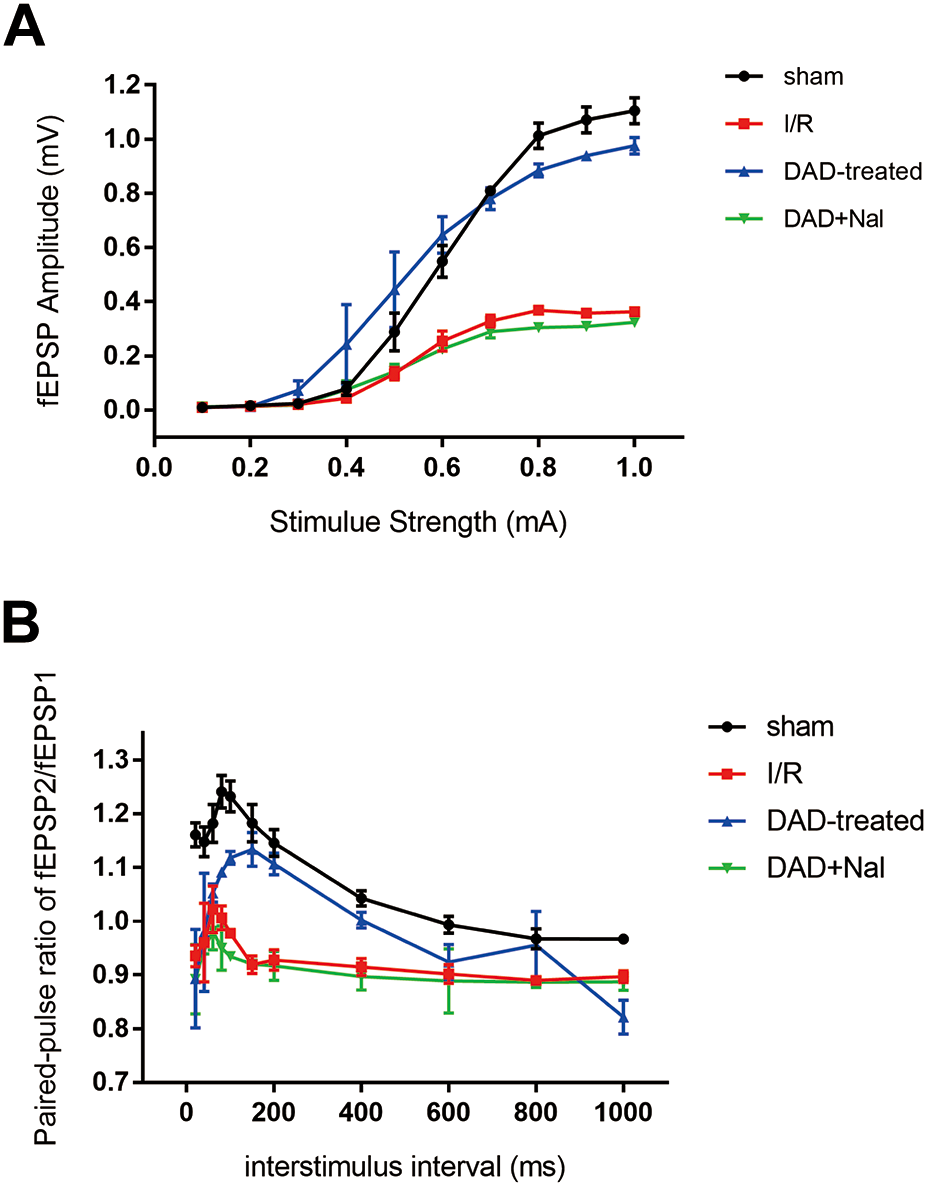
The changes of synaptic transmission in hippocampus on day 7 after global ischemia. (A) Input-output curve in the hippocampal slices on day 7 after global ischemia (N = 8 slices/4 rats). (B) Paired-pulse response in hippocampal slices on day 7 after global ischemia (N = 8 slices/5 rats).
PKCα/p-MARCKS Signaling in Response to the Morphological Plasticity of Hippocampal Neurons
To explore the molecular mechanism underlying the effects of DADLE on dendritic spine morphology (Figs. 3, 4), hippocampal PKCα/ε/γ and p-MARCKS were detected on days 3 and 7 post-ischemia. Although no changes were found in PKCε/γ (Fig. 6B, C), the PKCα levels had increased significantly in the rats on day 3, but decreased on day 7 after ischemia compared to the sham rats (Fig. 6A), suggesting that the specific subtype of PKC family plays different roles in global ischemia, but on the other hand, a differential endogenous PKCα response to various stages of I/R damage. Such changes in PKCα levels on days 3 and 7 post-ischemia were all reversed by DADLE treatment, which could be subsequently reversed by naltrindole (Fig. 6A). As a primary target of protein kinase C implicated in synapse formation and maintenance, the phosphorylation of MARCKS appeared to be coordinately influenced by changes in PKCα. The levels of p-MARCKS in the I/R group of rats were downregulated on day 3 and upregulated on day 7 after I/R injury (Fig. 6D). DADLE treatment exhibited a significant inhibitory effect on the phosphorylation of this protein in the DAD-treated group of rats on day 3 or day 7 post-ischemia, and the inhibitory roles of DADLE could be counteracted by naltrindole (Fig. 6D). These results suggest that DOR-mediated PKCα/p-MARCKS signaling might play an important role in the morphological plasticity of hippocampal neurons suffering from global ischemia.
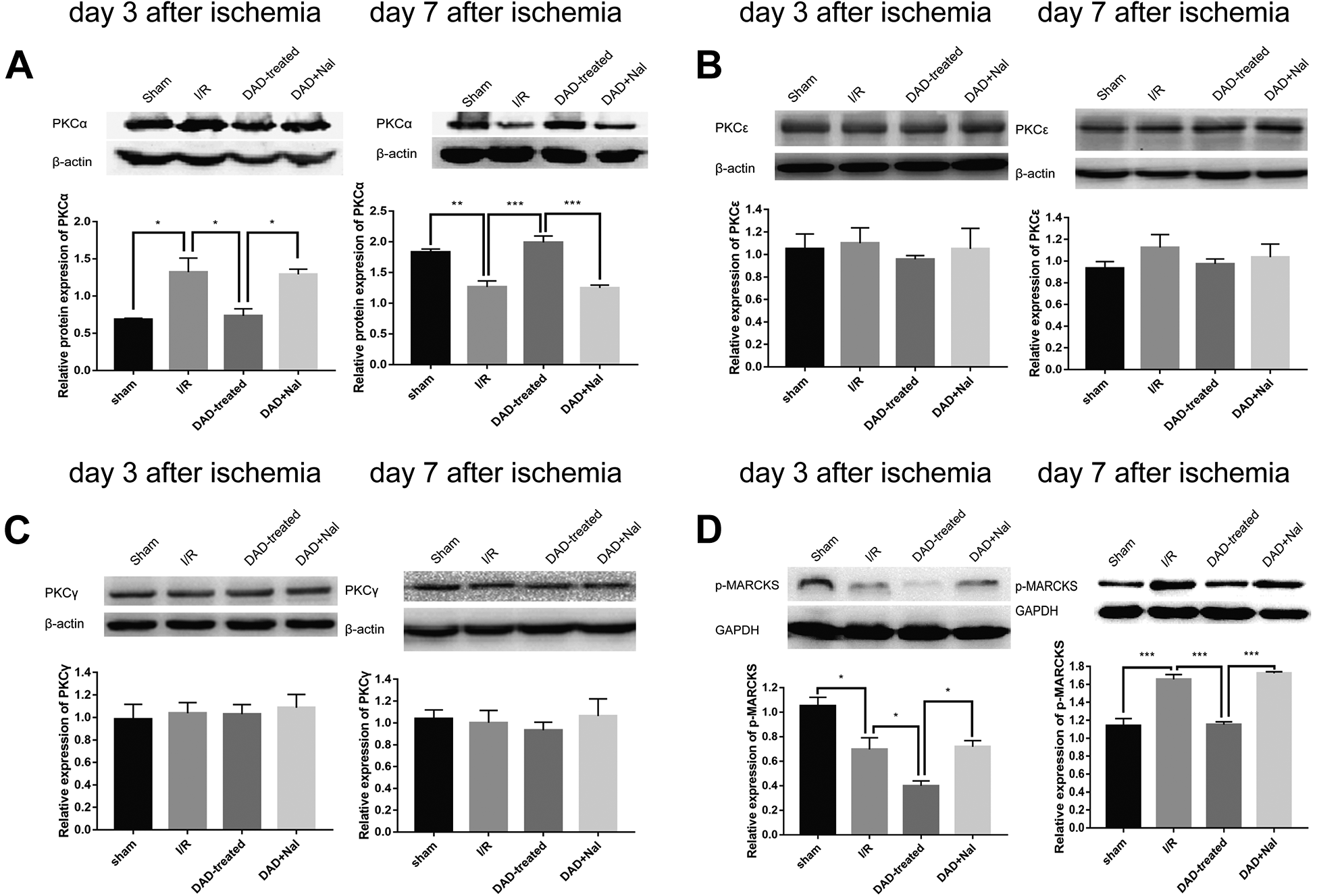
DOR activation with DADLE decreased p-MARCKS via PKCα, not PKCε/γ, after global ischemia. Western blot analysis of PKCα (A)/ε (B)/γ (C), p-MARCKS (D) expression in rat hippocampus on day 3(left) and day 7(right) after global ischemia. (N = 3 per group) *P < 0.05. **P < 0.01. ***P < 0.001.
BDNF/p-ERK1/2/p-Synapsin I Signaling in Response to the Transmission Plasticity of Hippocampal Neurons
To reveal the molecular responses underlying the beneficial effects of DOR activation on the synaptic transmission against I/R damage as shown in Fig. 5, hippocampal BDNF and its downstream signaling coordinators such as ERK1/2 and synapsin I were examined in the rats after 3 and 7 days of I/R injury, respectively. The results showed that BDNF expression was mildly elevated in the I/R group of rats on day 3, but was suppressed on day 7 post-ischemia as compared with the sham rats, indicating a time-related decline of the intrinsic adaptive function of this neurotrophic protein as I/R injury was prolonged (Fig. 7A). Remarkably, the levels of hippocampal BDNF increased significantly in the DAD-treated rats compared to the I/R rats on day 3 or day 7 post-ischemia, and the stimulating effects of DADLE on BDNF could be counteracted by naltrindole (Fig. 7A). Accordingly, the expression of p-ERK1/2 and p-synapsin I was altered in response to BDNF activity. The data showed that p-ERK1/2 levels were all reduced in the I/R rats, both on days 3 and 7 post-ischemia, which appeared to be neutralized in the rats with DADLE treatment (Fig. 7B). Such up-regulating roles of DADLE in mediating p-ERK1/2 activities were counteracted by naltrindole (Fig. 7B). The changes in p-synapsin I expression also exhibited a time-related interaction in the I/R rats of the ischemic insult with the endogenous defensive responses, showing an increase in its expression level on day 3 and a decrease in its expression on day 7 post-ischemia, which was reversed in the rats with DADLE treatment, and remained unchanged in the DAD+Nal group of rats, as compared with the rats in the I/R group (Fig. 7C). These results suggest that DOR activation could functionally modulate the pro-survival signaling pathway, that is, BDNF/p-ERK1/2/p-synapsin I, with particular implications for improving transmission plasticity against prolonged global ischemic injury.
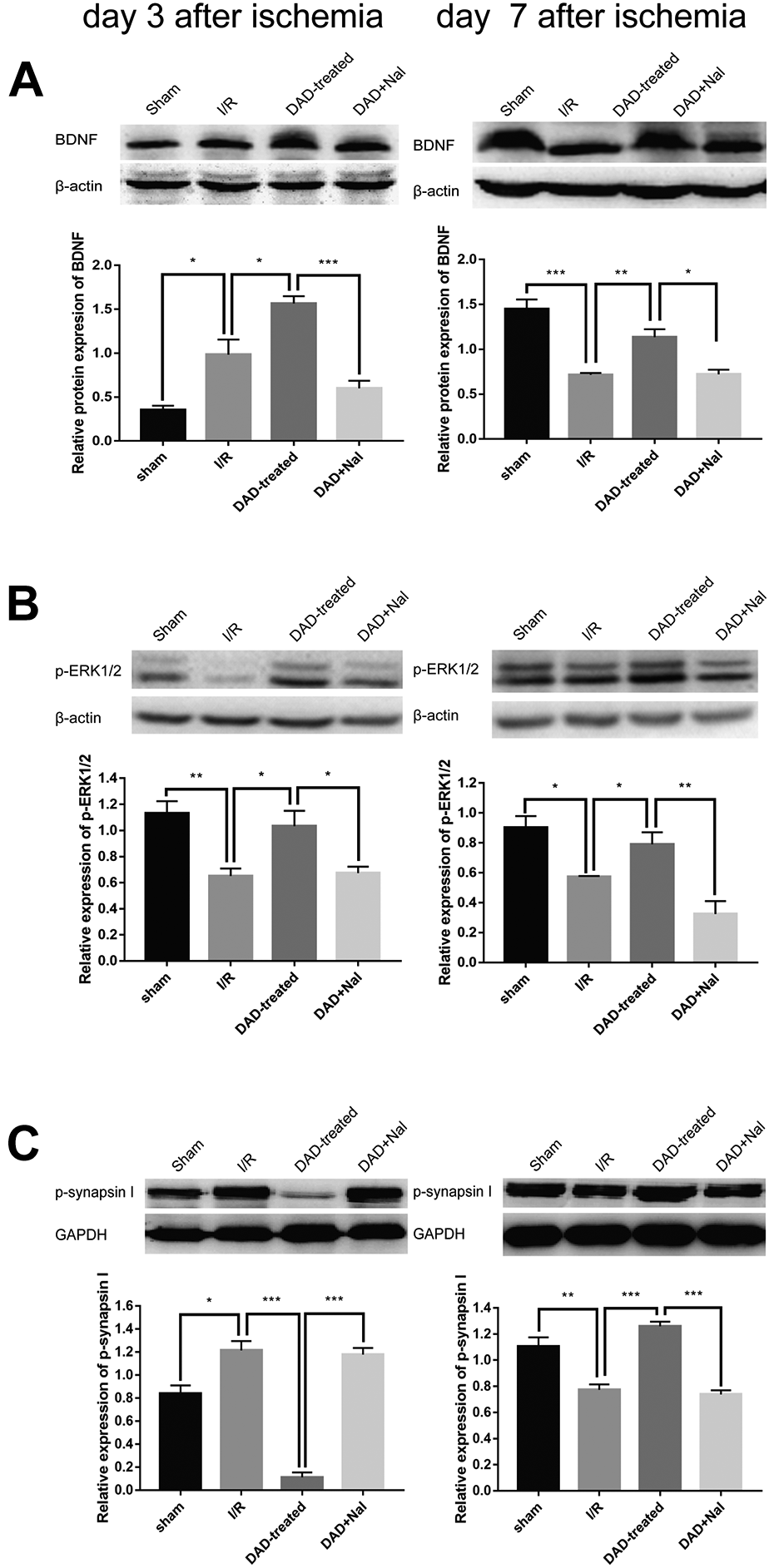
DOR activation with DADLE regulated p-synapsin I by BDNF-p-ERK pathway after global ischemia. Western blot analysis of BDNF (A), p-ERK1/2 (B), p-synapsin I (C) expression in rat hippocampus on day 3(left) and day 7(right) after global ischemia. (N = 3 per group) *P < 0.05. **P < 0.01. ***P < 0.001.
Discussion
The damaging events in global ischemic injury include acute injury (such as excitotoxicity and free radical injury) and prolonged damage (such as edema and inflammation), eventually leading to neuronal death after days of ischemia 3 . DADLE, a DOR agonist, has been proven to have therapeutic effects in stroke and many other neurological disorders including epilepsy, spinal cord injury, and neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease, with poorly understood mechanisms 25 . In this study, we showed that DADLE-induced DOR activation promoted CA1 neuronal survival and astrocytic activation on day 7 post-ischemia (Fig. 2, S1). These results were similar to our previous findings in rats with 3 days of I/R injury 23 , which further revealed that spatial memory defects were alleviated in rats with prolonged ischemic insult until day 9 21 . These findings were also similar to a previous report by Gao et al., in which DOR activation by hypoxic preconditioning attenuated the loss of CA1 neurons in the asphyxial cardiac arrest rat on day 7 of restoration 11 , suggesting that DOR activation-mediated neuroprotection would be long-lasting in combating ischemia-induced neuronal death.
Global ischemia may induce a progressive decrease in CA1 synapses and a rapid increase in perforated synapses 26 and may also lead to increased presynaptic [Ca2+]i and depressed evoked fEPSPs 27 . This suggests that DADLE-mediated alleviation of ischemia-induced memory defects may largely be attributed to recovery of synaptic morphology and function. Thus, we investigated synaptic alterations, both morphologically and functionally, of hippocampal neurons in rats on days 3 and 7 after ischemia with or without DADLE treatment. The results showed that DOR activation could significantly ameliorate not only the impaired synaptic structures, as evidenced by the increase in the density of dendritic spines (Figs. 3 and 4), but also damaged CA3-CA1 synaptic transmission, as indicated by the increase in fEPSP amplitude and PPR (Fig. 5). These results provide compelling evidence that suggests a synaptic mechanism of the action of DADLE-mediated DOR activation towards the amelioration of learning and memory in rats with global ischemia that we reported previously 21 .
Given the fact that the PKC and its substrate MARCKS signaling axis play crucial roles in the maintenance of dendritic spine morphology, we then detected the expression of PKC family and the status of MARCKS phosphorylation at both day 3 and 7 post-ischemia, based on the finding that neurons and astrocytes may have differential alterations at these two time points 28 . We found that 3-day ischemic stress resulted in an increase in PKCα but not PKCε/γ, while a decrease in p-MARCKS (Fig. 6), and DADLE treatment resulted in differential inhibition of PKCα and a further decline in MARCKS phosphorylation (Fig. 6A, D). This result suggested that ischemia-induced acute excitotoxicity activated downstream detrimental PKC signaling that was counteracted by DADLE treatment, and the reduced p-MARCKS we observed might be an endogenous protective action against ischemia, and the protective action was further strengthened by DADLE treatment. The response patterns of PKCα-MARCKS signaling were distinguishable on day 7 from that on day 3 post-ischemia, in which ischemia-induced PKCα declined while p-MARCKS increased, and the changes were all reversed by DADLE treatment (Fig. 6A, D), indicating that as the ischemic insult was prolonged, the detrimental effects of p-MARCKS became more severe and could be restored by DADLE-induced DOR activation. Previous studies have shown that MARCKS can be phosphorylated by PKC, leading to spine shrinkage or collapse 29 . Similarly, abnormal phosphorylation of MARCKS by overactivated PKC initiates synapse pathology in the early stage of Alzheimer’s disease 30 , and reduces synaptic complexity in cortical neurons with loss of the 22 γ-protocadherins (γ-Pcdhs) 31 . In contrast, MARCKS dephosphorylation was found to be involved in neurite outgrowth in neuroblastoma cells via bradykinin treatment 32 and insulin-like growth factor-I (IGF-I) stimulation 33 . Collectively, these results suggest that the neuronal PKCα-MARCKS signaling axis is closely involved in the maintenance of dendritic spine morphology and synapse integrity, and their activities, such as phosphorylation or dephosphorylation of MARCKS, need to be properly regulated when the central nervous system encounters an insult. Furthermore, there is evidence that alleviation of ischemic insult by OR activation is possibly through reduced PKC expression 34,35 , and the ubiquitination degradation of MARCKS may be involved in rapid ischemic tolerance following ischemic preconditioning 36 . The differential changes in PKCα and p-MARCKS on day 3 and day 7 post-ischemia and DADLE treatment found in this study further suggest that PKC and MARCKS may go beyond a simple kinase-substrate relationship 37 . Although PKCε/γ has also been reported to participate in neuroprotection in a focal ischemic mode l 38,39 , the expression of PKCε/γ did not change in the DADLE-treated group, suggesting that PKCα/ε/γ plays different roles in global and focal ischemic rats, and p-MARCKS may be associated, at least in part, with PKCα, but not PKCε/γ, in response to DOR neuroprotection against I/R insult. Clearly, the PKC family is subjected to complex regulation, not only by simple changes in protein content. The unique mechanisms between PKC subtypes warrant further investigation. The above results implied that PKCα/p-MARCKS signaling might be involved in the morphological improvement of ischemic synapses treated by DADLE-induced DOR activation.
BDNF-induced LTP was reported to be required for ERK activation in the rat hippocampus 40 . BDNF and its downstream signaling are also involved in the neuroprotective effects of (-)-phenserine against focal ischemic insult 41 . It has been documented that synapsin I may act as a downstream player in BDNF-mediated synaptic formation via ERK signaling 42 , and that phosphorylation of synapsin I is involved in the release of neurotransmitters in synaptic transmission 43 . To understand the possible interaction between DOR activation and BDNF signaling underlying the anti-ischemic effects of DADLE on synaptic transmission, we examined the changes in hippocampal BDNF and downstream components such as ERK1/2 and synapsin I in the rats on days 3 and 7 post-ischemia and DADLE treatment. The results showed that BDNF expression had a mild increase on day 3 and a decrease on day 7 post-ischemia, and its expression levels at both time points post-ischemia had all increased following DADLE treatment (Fig. 7A). Concordantly, p-ERK1/2 expression decreased significantly in the ischemic rats and was restored by DADLE treatment (Fig. 7B). Similar to PKCα-MARCKS signaling as described above, the changes in BDNF on day 3 post-ischemia appeared to be an endogenous adaptive reaction to the ischemia-induced excitotoxicity, which in turn faded out on day 7 post-ischemia. In contrast, the expression of p-ERK1/2 remained lower, indicating an inhibition of the pro-survival signaling pathways that might promote neuronal death and thereby be detrimental to hippocampal synaptic transmission (Fig. 5). Furthermore, the differential changes of p-synapsin I in response to ischemic insults as well as DADLE treatment were in accordance with those of BDNF-p-ERK1/2. As shown in Fig. 7C, p-synapsin I was significantly increased on day 3 and reduced on day 7 post-ischemia, suggesting a cross-talk of this protein with the coexisting endogenous defense featured by the mild induction of BDNF and the neuronal deterioration characterized by the reduction of p-ERK1/2 at an earlier stage of ischemic insult (day 3) and a consequence responsible for the functional inhibition of both BDNF and ERK1/2 at the advanced stage of ischemic damage (day 7). Identical to the changes in BDNF-ERK1/2 signaling as described above, the differential expression of p-synapsin I was also functionally rescued by DADLE (Fig. 7C). Thus, our data provide preliminary evidence that BDNF/p-ERK1/2/p-synapsin I signaling may be involved in the impairment of hippocampal synaptic transmission and contribute to cognitive dysfunction in prolonged (up to day 7) ischemia-reperfusion injury. Meanwhile, the prolonged detrimental effects of cerebral ischemia were all counteracted by DADLE treatment, while all of these pharmacological efficacies of DADLE were eliminated by Naltrindole (Figs. 5, 7), indicating that DOR activation may be responsible for BDNF/p-ERK1/2/p-synapsin I signaling pathway-mediated neuronal transmission recovery from cognitive decline in rats with cerebral ischemia.
In summary, the present study demonstrated that DOR activation-induced protective signaling pathways of PKCα-MARCKS in synaptic morphology and BDNF-ERK-synapsin I in synaptic transmission toward cognitive recovery in ischemic rats. It is still worth considering the coordination of both signaling pathways, rather than merely individual ones, in improving synaptic structure and/or function. Indeed, several lines of evidence have revealed that PKCα facilitates synaptic plasticity with the help of BDNF signaling 44 , suggesting that DADLE-induced upregulation of PKCα on day 7 post-ischemia may also be associated with BDNF signaling (Fig. 6A). The PKC/ERK signaling pathway has also been implicated in DOR neuroprotection in an in vitro model 45 , suggesting the possibility that DADLE-induced elevation of PKCα and BDNF may participate in the activation of ERK1/2 on day 7 post-ischemia (Figs. 6A, 7). These findings indicate an interaction between PKCα-MARCKS and BDNF-ERK-synapsin I in protecting synaptic plasticity against cerebral ischemia.
Supplemental Material
Supplemental Material, sj-docx-1-cll-10.1177_09636897211041585 - Delta Opioid Receptor Activation with Delta Opioid Peptide [d-Ala2, d-Leu5] Enkephalin Contributes to Synaptic Improvement in Rat Hippocampus against Global Ischemia
Supplemental Material, sj-docx-1-cll-10.1177_09636897211041585 for Delta Opioid Receptor Activation with Delta Opioid Peptide [d-Ala2, d-Leu5] Enkephalin Contributes to Synaptic Improvement in Rat Hippocampus against Global Ischemia by Guangming Zhang, Zelin Lai, Lingling Gu, Kejia Xu, Zhenlu Wang, Yale Duan, Huifen Chen, Min Zhang, Jun Zhang, Zheng Zhao and Shuyan Wang in Cell Transplantation
Supplemental Material
Supplemental Material, sj-jpg-1-cll-10.1177_09636897211041585 - Delta Opioid Receptor Activation with Delta Opioid Peptide [d-Ala2, d-Leu5] Enkephalin Contributes to Synaptic Improvement in Rat Hippocampus against Global Ischemia
Supplemental Material, sj-jpg-1-cll-10.1177_09636897211041585 for Delta Opioid Receptor Activation with Delta Opioid Peptide [d-Ala2, d-Leu5] Enkephalin Contributes to Synaptic Improvement in Rat Hippocampus against Global Ischemia by Guangming Zhang, Zelin Lai, Lingling Gu, Kejia Xu, Zhenlu Wang, Yale Duan, Huifen Chen, Min Zhang, Jun Zhang, Zheng Zhao and Shuyan Wang in Cell Transplantation
Footnotes
Abbreviations
Author Contributions
GMZ, ZLL, and LLG carried out experiments and wrote the manuscript. ZLW, HFC, YZL, and KJX carried out experiments and analyzed experimental results. MZ and YLD assisted with data analysis. YSW, ZZ, and JZ designed and instructed experiments. All authors contributed to and approved the final manuscript.
Ethics approval
This study was approved by the Institutional Animal Care and Use Committee of East China Normal University.
Statement of Human and Animal Rights
All experiments involving animals were performed in accordance with Animals Act (2006, China) and approved by the Institutional Animal Care and Use Committee of East China Normal University (AR201404022).
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by grants from the National Natural Science Foundation of China (81301122 to SW, 31171019 to ZZ, and 81470829 to JZ), the Key projects of Science and Technology Committee of Changning District, Shanghai (CNKW2017Z01 to GZ) and an opening grant from Key Laboratory of Brain Functional Genomics (ECNU), Ministry of Education (JZ).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.