
Abstract
We examined the functionality of hippocampal CA1 neurons at early times after transient global ischemia, by electrophysiologic recordings in brain slices. Transient ischemia was conducted on rats using the method of 15-minute four-vessel occlusion, and brain slices were obtained from these animals at different times after ischemia. Within 24 hours after insult, CA1 neurons showed no substantial damage as identified by morphologic means, but exhibited dramatic decreases in synaptic activities by 12 hours after insult, which became further decreased at more extended times after recovery. Blocking γ-aminobutyric acid A (GABAA) receptors with bicuculline produced a reversible augmentation of the diminished synaptic responses in slices prepared from 12-hour postinsult animals, but failed to do so in slices obtained from rats 24 hours after insult. Recorded in whole-cell mode, the minimum depolarizing current required to elicit an action potential was about twofold larger in the ischemic CA1 neurons than in sham controls, suggesting that an elevated spiking threshold exists in these neurons. We suggest that decreases in electrophysiologic activities precede the morphologic deterioration in postischemic CA1 neurons. The early decrease in CA1 synaptic activities may be associated with an imbalance between glutamate-mediated synaptic excitation and GABAA-mediated synaptic inhibition, whereas substantial impairments in synaptic transmission likely take place after prolonged postischemic recovery.
In both humans and animal models, transient cerebral ischemia causes delayed neuronal death, which occurs a few days after the insult, depending on the severity of the ischemia (Pulsinelli et al., 1982; Kirino, 1982; Zola-Morgan et al., 1986, 1992). This delayed neuronal death has been observed in multiple areas of the brain (Rod and Auer, 1992); however, neurons in the CA1 region of hippocampus have been noted to be highly vulnerable in both humans and animal models. It is generally thought that the ischemic insult promotes an overstimulation of glutamate receptors, which leads to an elevation of intracellular Ca2+ (Choi, 1994; Desphande et al., 1987; Dienel, 1984; Olney, 1978). Treatments such as a blockade of glutamate receptors and the chelation of intracellular calcium during or shortly after the insult have been found to be protective against ischemic damage in some focal (Sheardown et al., 1990; Tymianski et al., 1993) and global ischemia animal models (Buchan et al., 1991b; Gill and Woodruff, 1990; Li and Buchan, 1993; Nellgard and Wielock, 1992; Rod and Auer, 1992; Sheardown et al., 1993). Mechanisms secondary to these initial events are likely involved, however, because the vulnerable CA1 neurons initially recover after the transient insult, and then slowly degenerate over a few days. Currently, the pathologic processes that lead to the delayed neuronal degeneration after transient ischemia are not fully understood.
In the past, many studies have characterized morphologic features of the delayed neuronal death; however, limited information is available regarding the functionality of the vulnerable neurons during postischemic recovery (Suzuki et al., 1983; Petito and Pulsinelli, 1984; Urban et al., 1989; Jensen et al., 1991). Studying the pathophysiologic features of postischemic CA1 neurons may provide new insights into the mechanisms underlying the delayed neuronal degeneration. In the current experiments, we examine the electrophysiologic properties of rat hippocampal CA1 neurons after transient ischemia, particularly within the first 24 hours of postischemic recovery. We asked the following questions: Do CA1 hippocampal neurons display altered functionality at early times after transient ischemia? If so, what is the conferred electrophysiologic feature, and what is the time course through which this dysfunction develops?
To address these questions, we created transient global ischemia in rats using the 15-minute four-vessel occlusion method, and then allowed the animals to recover for different times after the insult. At specific times, brain sections were obtained from ischemic animals for morphologic and electrophysiologic assessments. We demonstrate here that within 24 hours of postischemic recovery, synaptic responses resulting from Schaffer collateral stimulation are dramatically decreased well before any morphologically identified CA1 neuronal injury is observed. Furthermore, the threshold for action potential generation in postischemic CA1 neurons is elevated in a time-dependent manner after the ischemic insult, indicating that a down-regulation in CA1 neuronal activity precedes neuronal expiration.
METHODS
Transient global ischemia was produced in adult male Wistar rats (200 to 350 g) using the method established by Pulsinelli and Brierley (1979) with minor modifications. Briefly, on day 1, rats were anesthetized with 1.5% halothane in a 2:1 nitrous oxide–oxygen mixture, and the vertebral arteries were electrocauterized through the alar foramina. Both carotid arteries were isolated and encircled by loose ligatures. Twenty-four hours later, one femoral artery was catheterized under halothane anesthesia (0.8%) to monitor blood gases and arterial blood pressure. After discontinuing halothane, both carotid arteries were clamped with aneurysm clips for 15 minutes. Blood gas measurements were done 10 minutes before applying clips, and 1 minute before and 10 minutes after removing clips. Rectal temperature was maintained at 36.5 to 37.5°C throughout the experiment with a warming lamp. Morphologic and electrophysiologic examinations were performed only if animals showed an isoelectric EEG during the entire occlusion period and recovered EEG activity within 30 minutes after the occlusion. The blood gas parameters of these animals were in the following range: 7.30 < pH < 7.55, 25 < P
For morphologic examinations, rats were anesthetized with halothane and perfused with a 4% phosphate-buffered formalin solution by transcardiac puncture. After perfusion, the brain was removed and left in a 10% phosphate-buffered formalin solution (pH 7.4) overnight. Sacrifice was done at these different in vivo reperfusion times: 3 hours, 24 hours, 3 days, and 7 days. Coronal brain sections of 6 μm were cut at five specific septotemporal levels identified by their configuration and stained with cresyl violet (Smith et al., 1984; Rod and Auer, 1992). We quantified neuronal injury by direct light microscopic examination on these sections. CA1 neurons were scored as injured if they displayed shrunken, irregular cell bodies with darkly stained granules (Kirino et al., 1985). At each level, three to four sections were examined, and the number of injured CA1 neurons was divided by the total number of the neurons and multiplied by 100 to give the percent neuronal damage. Mean values for each animal were included in data analysis. Analysis of variances on ranks (Dunn's method) was used to detect statistical difference.
Brain slices for electrophysiologic recordings have been previously described (Zhang et al., 1991). Slices were taken from rats after in vivo reperfusion of 1, 6, 12, and 24 hours, and 6 to 7 days, respectively. The rats were anesthetized with halothane and decapitated immediately. The brain was removed and maintained in an ice-cold artificial cerebrospinal fluid (ACSF) for 3 to 5 minutes before slicing. The hemisectioned brain was mounted on an aluminum block and sliced in the ice-cold ACSF using a vibrotom (series 1000, Technical Products International Incorporated, St. Louis, U.S.A.). Eight to nine transverse brain slices of 400-μm thicknesses were obtained from each half-brain. Slices then were kept in oxygenated ACSF at room temperature for at least 1 hour before recording and not more than 7 hours after dissection. The components of the ACSF are (in mmol/L): NaCl 125, KCl 2.5, NaH2PO4 1.25, CaCl2 2, MgCl2 1.3, NaHCO3 26, and glucose 10. When aerated with 5% CO2−95% O2, the pH value of the ACSF is 7.4. Osmolarity of the ACSF is 300 ± 5 mOsm.
For electrophysiologic recordings, slices were transferred to a fully submerged chamber and perfused with the oxygenated ACSF continuously. The perfusion rate was 4 to 5 mL/min. We also applied humidified, warmed 5% CO2−95% O2 over the perfusate to ensure a warm, oxygenated local environment. All recordings were performed at slice temperatures of 33° to 34°C. Field potentials were recorded extracellularly through glass pipettes filled with 150 mmol/L NaCl, and the electrode was placed in the somatic or apical dendritic layer of the CA1 region. Electrical stimulation of Schaffer collateral afferents was performed using a bipolar tungsten electrode placed in the stratum radiatum at the CA1−CA2 border. Constant current pulses of 0.1- to 0.2-millisecond durations were generated through a Grass S88 stimulator and delivered through an isolation unit every 15 seconds. Data were collected only if stable field potentials were recorded, and averages were obtained from 5 to 10 recordings. Because a submerged chamber and bipolar stimulating electrode were used, the current pulses released from the stimulation isolation unit may not represent the actual current passing through slice tissue because of current shunting. Thus, we normalized the stimulation strength as multiple times of the threshold intensity, and this normalized stimulation strength was used to generate the stimulation–response plot. To examine the paired pulse facilitation (PPF) in dendritic responses, two identical stimulating current pulses at 50% of maximal intensity were delivered with a separation interval of 50 milliseconds. The PPF was determined by measuring the percentage increase in the peak amplitude of the second response, taking the first response as 100%.
For whole-cell patch recordings, we used a standard solution to fill patch pipettes, and this solution contained the following in millimoles: 150 potassium methylsulfate, 2 HEPES, 0.5 ATP, and 0.1 EGTA (Zhang et al., 1994). In some experiments, 1 to 1.5 mmol/L BAPTA or 4,4′-difluoro BAPTA was added to the standard internal solution to improve the seal formation. The calculated dissociation constant values are 220 nmol/L and 1.7 μmol/L for BAPTA and difluoro BAPTA, respectively (Adler et al., 1991; Zhang et al., 1995). All patch pipette solutions had a pH of 7.25 adjusted with potassium hydroxide and osmolarity of 280 ± 10 mOsm. When filled with these solutions, the patch pipette had resistances from 3 to 5 MΩ. An Ag–AgCl reference electrode was placed in an agar–saline bridge to attenuate junctional potentials (Zhang et al., 1991; Zhang and Krnjević, 1993).
Electrical signals were recorded using an Axon-clamp amplifier 2A (Axon Instrument, Foster City, CA, U.S.A.), and data were stored and analyzed with Pclamp software (version 5.5, Axon Instrument). Input resistance was estimated by passing square current pulses (duration of 500 milliseconds to 1.0 second) at low intensities (30 to 50 picoamperes), and measurements were taken at the steady state of the voltage responses. The peak amplitude and the duration to half-maximal amplitude of the action potential were measured. To activate the slow afterhyperpolarization maximally, a train of action potentials of about 10 spikes (Zhang et al., 1994, 1995) was generated by depolarizing current pulses (200 to 300 milliseconds, 2 to 3 nanoamperes) at membrane potentials near −60 mV. The amplitude of the slow afterhyperpolarization was measured at 200 milliseconds after the end of the depolarizing pulse.
All solutions were made with deionized sterile water (pH 5 to 6, resistance 18.2 MΩ/cm) from a Milli-Q UV plus system. Chemicals for making the patch pipette solutions were purchased from Fluka (New York, New York, U.S.A.), except potassium methylsulfate (ICN, New York, New York, U.S.A.). BAPTA derivatives (potassium salt) were purchased from Molecular Probes (Eugene, OR, U.S.A.). Other drugs were purchased from Sigma (St. Louis, MO, U.S.A.).
RESULTS
Morphologic examinations
The morphologic features of CA1 neurons after ischemia were examined by direct light microscopic visualization at five specific septo-hippocampal levels (Smith et al., 1984; Rod and Auer, 1992; see Methods). The percentage of injured neurons is summarized in Fig. 1. Less than 6% of CA1 neurons were morphologically damaged when examined at 3 or 24 hours after reperfusion (Fig. 1). CA1 neuronal injury became pronounced at 3 days after reperfusion, and more than 90% of the CA1 neurons were injured at 7 days after reperfusion. These results are consistent with previous studies using similar transient global ischemic paradigms (Pulsinelli and Brierley, 1979; Rod and Auer, 1992; Smith et al., 1984).
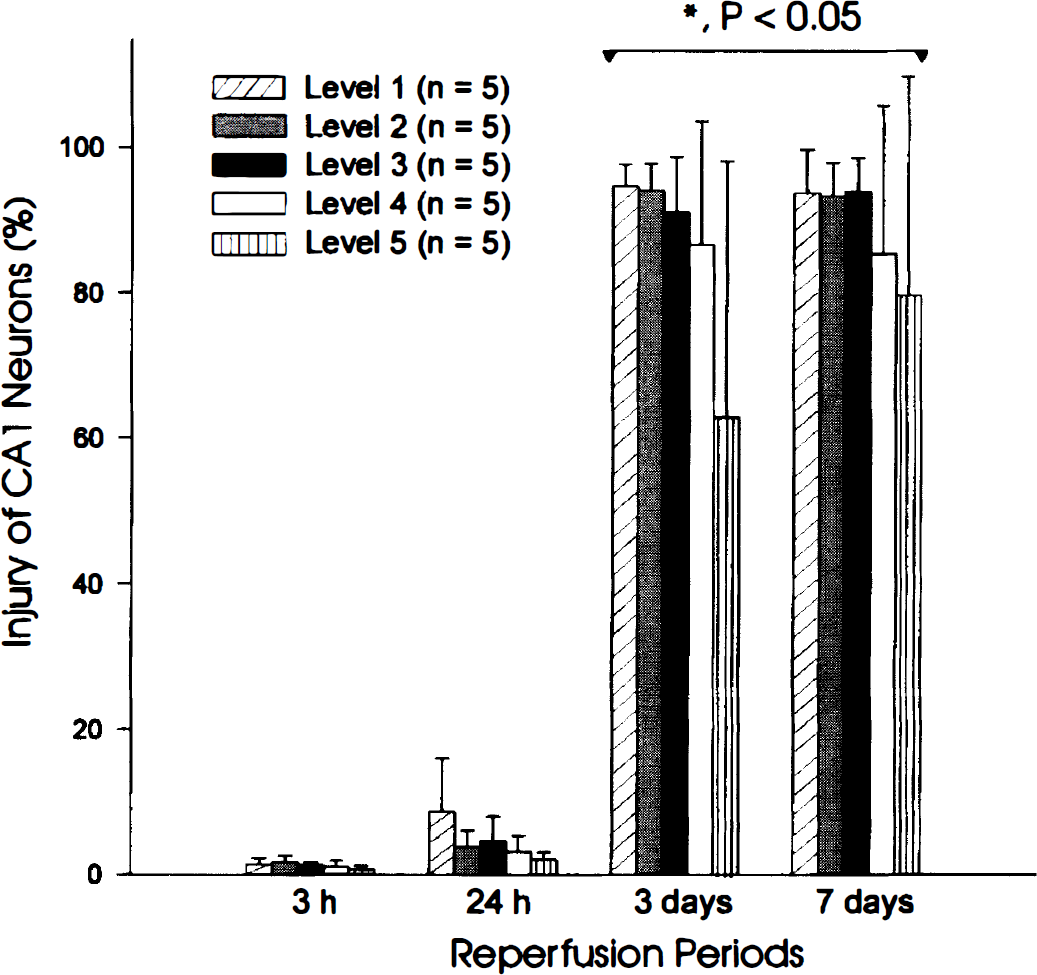
Morphologic examination of injured hippocampal CA1 neurons after transient global ischemia. The ischemic insult was conducted using the 15-minute four-vessel occlusion method, and the animals were allowed to recover for different times after reperfusion. At different reperfusion times as indicated, animals were sacrificed, and coronal brain sections of 6 μmol/L were cut at five specific septal levels and stained with cresyl violet (see Methods). At each level, three to four adjacent sections were examined, and data were collected from five animals for each time point after postischemia reperfusion. Neuronal injury was assessed by direct visualization under a light microscope. To calculate the percentage change, the number of injured CA1 neurons was divided by the total number of neurons examined at each level. Analysis of variance on ranks (Dunn's method) were used to detect significance from sham controls (P < 0.05). Notice the large proportion of cell injury observed after 3 days of reperfusion.
Electrophysiologic studies in brain slices
Sham-controlled slices were obtained at 24 hours (six rats) and 48 hours (three rats) after surgery. No change in any response examined was observed between sham animals of either postsurgery recovery times. Postischemic slices were taken after in vivo reperfusion of 1 hour (two rats), 6 hours (three rats), 12 hours (four rats), 24 hours (nine rats), and 6 to 7 days (two rats). To avoid potential differences in regional hippocampal responses (Ashton et al., 1989), we restricted our recordings to slices obtained from the dorsoseptal portion of the hippocampus. In the following text, we refer to each group according to their recovery time after surgery or ischemia. In whole-cell experiments performed on sham and 6-hour postischemia CA1 neurons, data were collected from two to four rats for each time point. Each recording was carried out on separate slices, and more than three CA1 neurons were collected per rat. Because of the difficulty in seal formation when performing whole-cell recordings on CA1 neurons 12 to 24 hours after ischemia (see later), one or two neurons were recorded from each slice, and data were collected from four to seven animals for 12- or 24-hour postischemia time points. No intention was made in the current experiments to compare the electrophysiologic parameters of CA1 neurons between sham and naive groups, and the sham responses presented here are used as standards to control for changes induced by surgery or anesthesia.
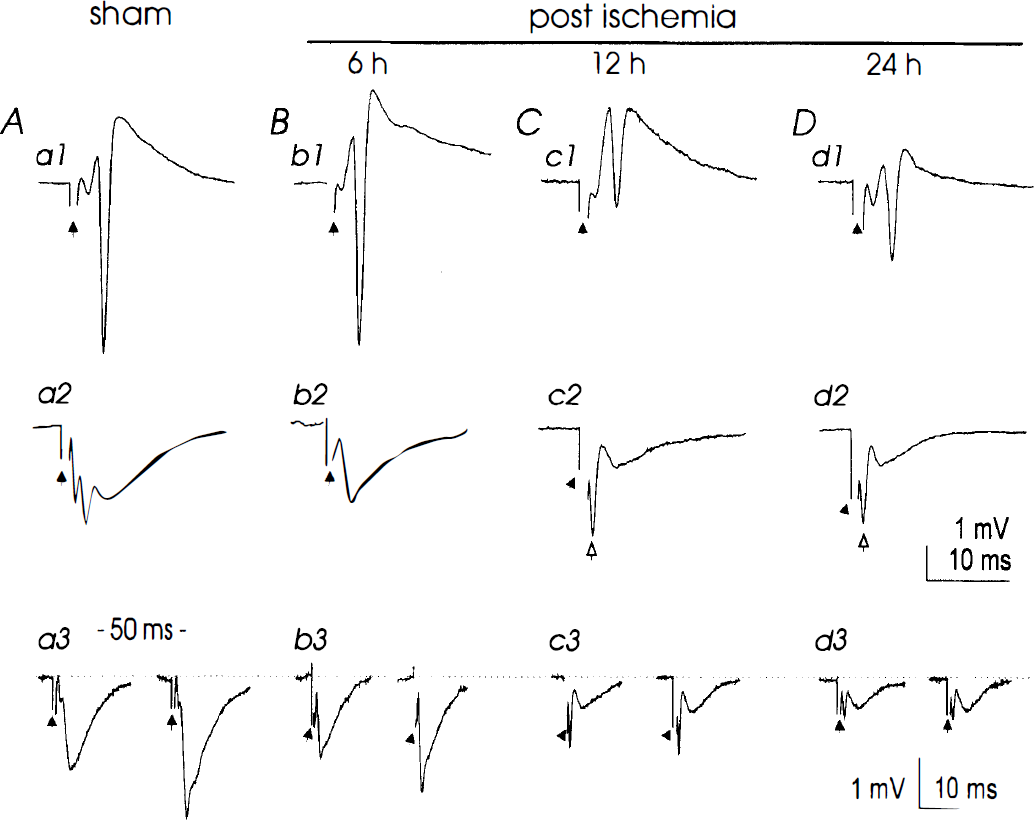
Decreased synaptic field potentials in the hippocampal CA1 region after ischemia. Representative records were collected from four individual slices prepared at 24 hours after surgery
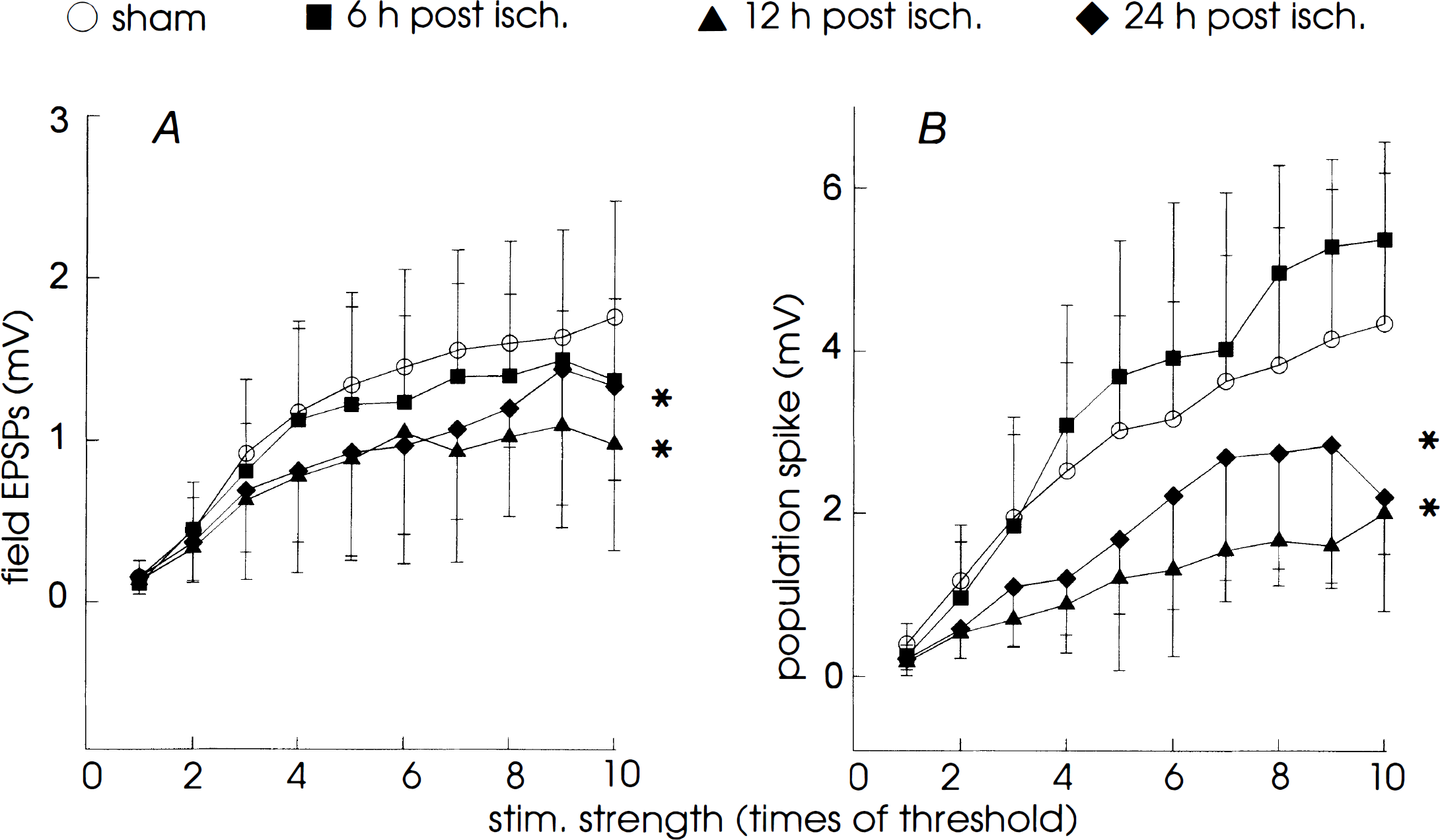
Stimulation–response relation of synaptic field potentials in sham control and postischemic slices. Amplitudes of dendritic field EPSP
Decreases in CA1 synaptic field potentials after transient ischemia
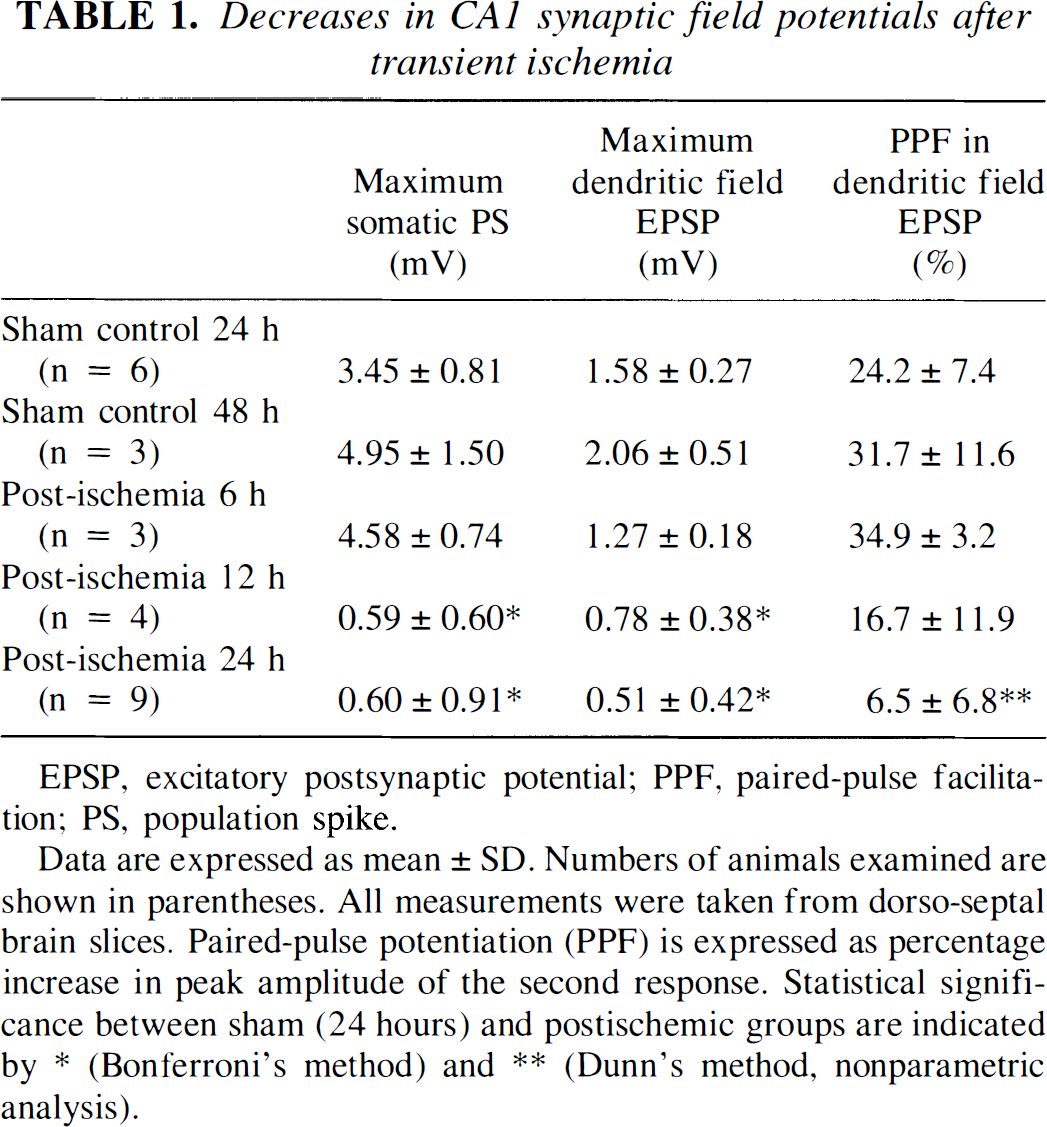
EPSP, excitatory postsynaptic potential; PPF, paired-pulse facilitation; PS, population spike.
Data are expressed as mean ± SD. Numbers of animals examined are shown in parentheses. All measurements were taken from dorso-septal brain slices. Paired-pulse potentiation (PPF) is expressed as percentage increase in peak amplitude of the second response. Statistical significance between sham (24 hours) and postischemic groups are indicated by * (Bonferroni's method) and ** (Dunn's method, nonparametric analysis).
Postischemia slices. Graded synaptic responses were consistently recorded from the CA1 region in slices obtained 6 hours after reperfusion (Fig. 2, b1, b2). The maximal amplitudes of the somatic and dendritic responses and the stimulation–response relations were comparable with those observed in sham groups (Table 1). Neither spontaneous activity nor a time-dependent deterioration in the synaptic field potentials was observed in these slices maintained in the ACSF for up to 7 hours, which is consistent with the observations obtained in sham slices. Because decreases in CA1 synaptic field potentials were readily detected in slices prepared 12 hours after in vivo reperfusion (see later), these results indicate that impairments in CA1 electrophysiologic responses do not evolve after incubation of slices in the isolated, low-temperature in vitro condition.
Synaptic field potentials with varied amplitudes were observed in slices prepared from ischemic animals after 12 and 24 hours of in vivo reperfusion. Strikingly, in about half of the slices examined (13 of 25 and 23 of 51 slices for the 12- and 24-hour groups, respectively), we observed no somatic PS after a single, maximal stimulus of Schaffer collateral afferents (Fig. 4, a1, b1). In the remaining slices, PS were observed, but these events displayed a small amplitude (Fig. 2, c1), or a lack of an initial upward waveform (Fig. 2, d1). No correlation was noticed between the inability to fire PS and the time the slices were maintained in ACSF. As shown in Table 1, the maximal amplitude of the somatic PS in postischemic slices was significantly smaller than that observed in sham controls. Similar decreases also were found in the dendritic field EPSP (Fig. 2, c2, d2) and the PPF (Fig. 2, c2, d2; Table 1). We generated a stimulation–response plot by collecting data from a group of slices that showed graded field EPSP (Fig. 3, filled triangles and diamonds). The normalized responses were significantly smaller in these ischemic slices than those obtained from sham controls at a wide range of stimulating strengths, suggesting decreased synaptic efficacy exists in most of the Schaffer collateral–CA1 synapses after prolonged reperfusion.
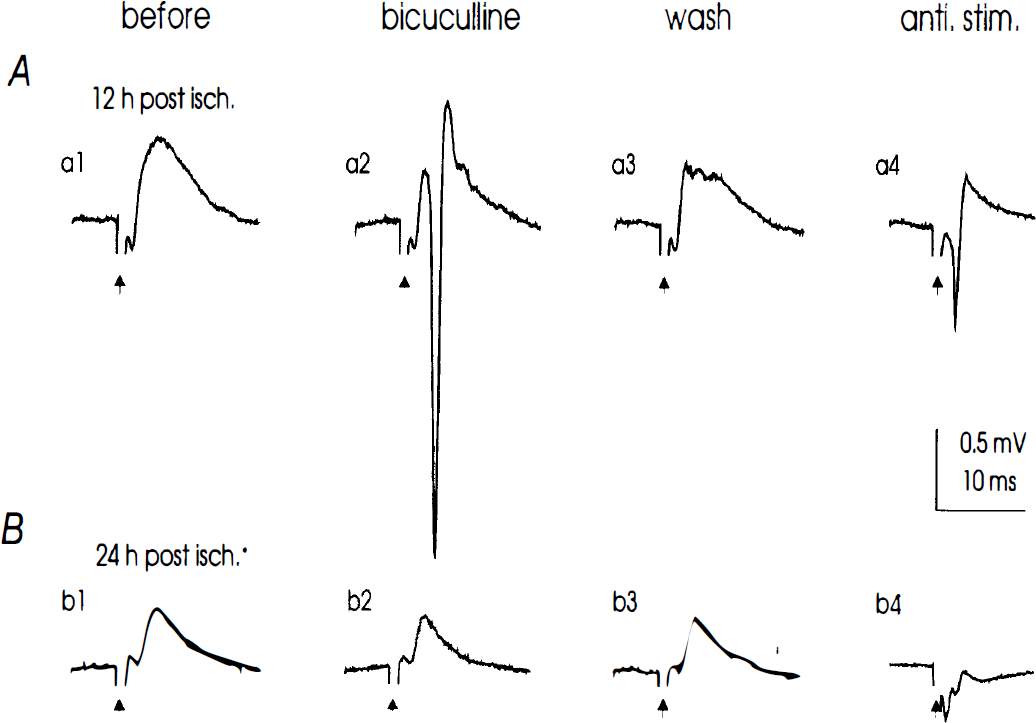
Effects of bicuculline methiodide (BMI) on somatic synaptic field potentials recorded from postischemic slices. Records were collected form two slices prepared at 12
Paired-pulse facilitation. To examine the plasticity in synaptic transmission, we used a PPF paradigm by stimulating Schaffer collateral afferents with twin stimuli separated by 50 milliseconds (see Methods). Previous studies demonstrate that multiple presynaptic mechanisms are involved in the PPF, including an inhibition of γ-aminobutyric acid (GABA) release by activation of presynaptic GABAB receptors (Nathan and Lambert, 1991) and direct enhancement of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor-mediated responses (Clark et al., 1994), likely through an elevation of basal intracellular Ca2+ in presynaptic terminals (Wu and Saggau, 1993). Thus, the PPF represents a convenient experimental protocol to test the function of Schaffer collateral afferents in addition to the amplitudes of CA1 field responses. In slices prepared 6 hours after reperfusion, the dendritic PPF in the CA1 region was comparable with that measured from sham controls (Fig. 2, a3, b3; Table 1). Significant decreases (P < 0.05) in the CA1 PPF were observed in slices obtained at 12 and 24 hours after reperfusion (Fig. 2, c3, d3; Table 1). These results suggest that the presynaptic mechanisms that promote synaptic excitation in sham Schaffer collateral–CA1 synapses are suppressed after prolonged postischemia reperfusion.
Effects of the GABAA receptor antagonist bicuculline. The lack of orthodromic PS in some postischemic slices prompted us to investigate the level of GABAA-mediated inhibition in these slices, since a strong GABAA-inhibition over glutamate excitation reduces the likelihood of neuronal firing. Thus, we examined the effect of bicuculline, a selective GABAA receptor antagonist, on the CA1 somatic field responses to reveal the potential synaptic excitation after the blockade of the GABAA-mediated inhibition. For comparison, only slices that failed to show a somatic PS after a maximal stimulus of Schaffer collateral afferents were treated with bicuculline (10 μmol/L, for 5 to 6 minutes). In 8 of 12 slices prepared from four rats at 12 hours after ischemia, orthodromic PS, which were not observed before treatment (Fig. 4, a1), were reversibly induced after application of bicuculline (Fig. 4, a2); however, the similar application of bicuculline failed to restore somatic PS in any slice (7 of 7) prepared from three rats at 24 hours after ischemia (Fig. 4, b1, b2).
Antidromic stimulation. In slices prepared 24 hours after reperfusion, the failure to restore CA1 PS after bicuculline application could result from decreased intrinsic excitability in postsynaptic CA1 neurons. To address this possibility, we recorded somatic responses after antidromic stimulation of CA1 axons in the Alveus. In five of five slices prepared from two animals after 12 hours of reperfusion, somatic PS were not induced after the orthodromic stimulation of Schaffer collateral afferents, but were clearly elicited after antidromic stimulation of the Alveus (Fig. 4, a4). The peak amplitude of the antidromic PS was 1.3 ± 0.3 mV. In similar experimental settings, antidromic PS were induced in only 3 of 14 slices prepared from four rats after 24 hours of reperfusion (Fig. 4, b4). These observations, taken together with the differential effects of bicuculline mentioned earlier and an elevation of spiking threshold (see later), strongly suggest that the intrinsic excitability of CA1 neurons is decreased after 24 hours of postischemic reperfusion.
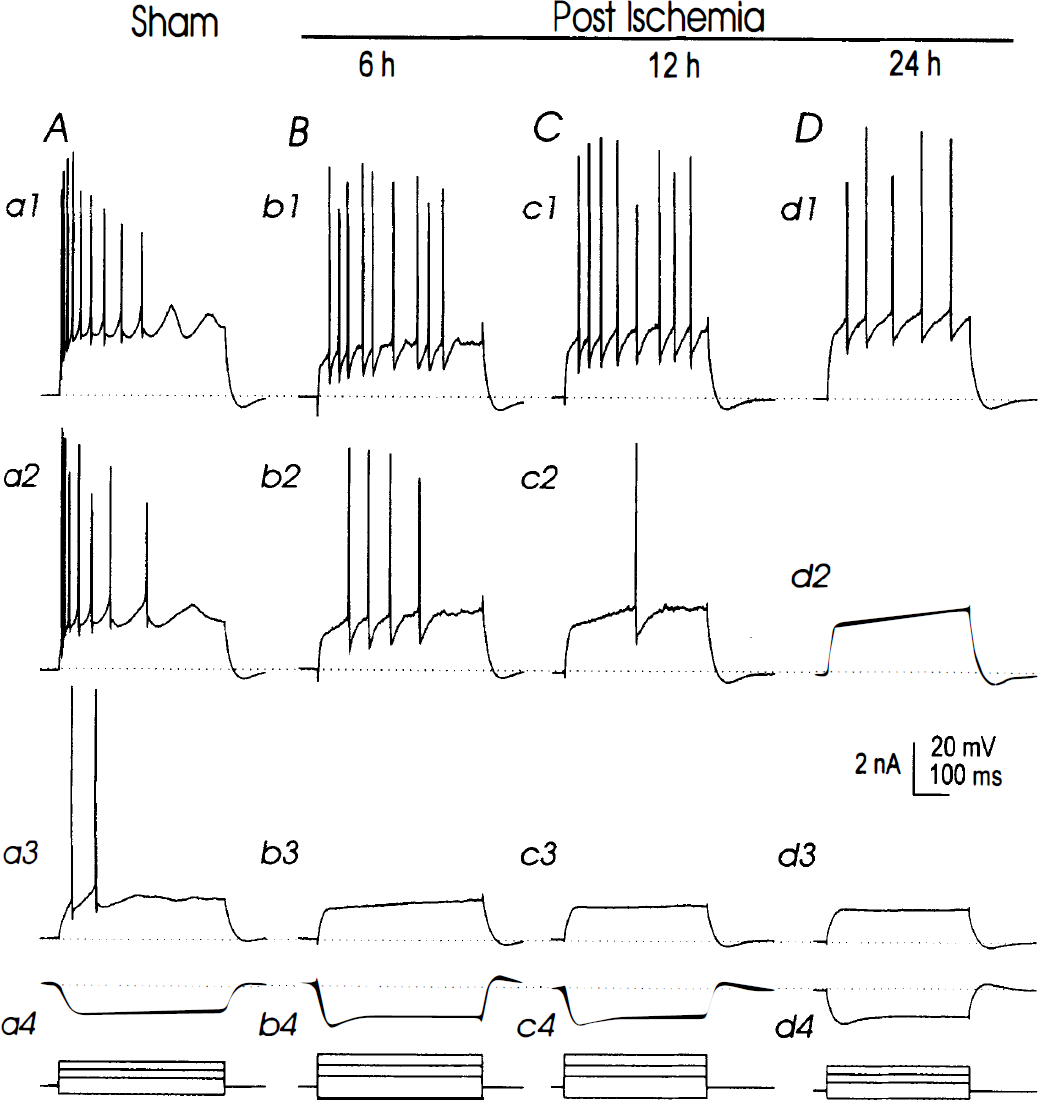
Increased firing threshold and decreased firing adaptation in postischemic CA1 neurons. Records were collected from four individual neurons in slices prepared 24 hours after sham surgery
Electrophysiological properties of sham and postischemic CA1 neurons recorded in whole-cell mode (mean ± SD)
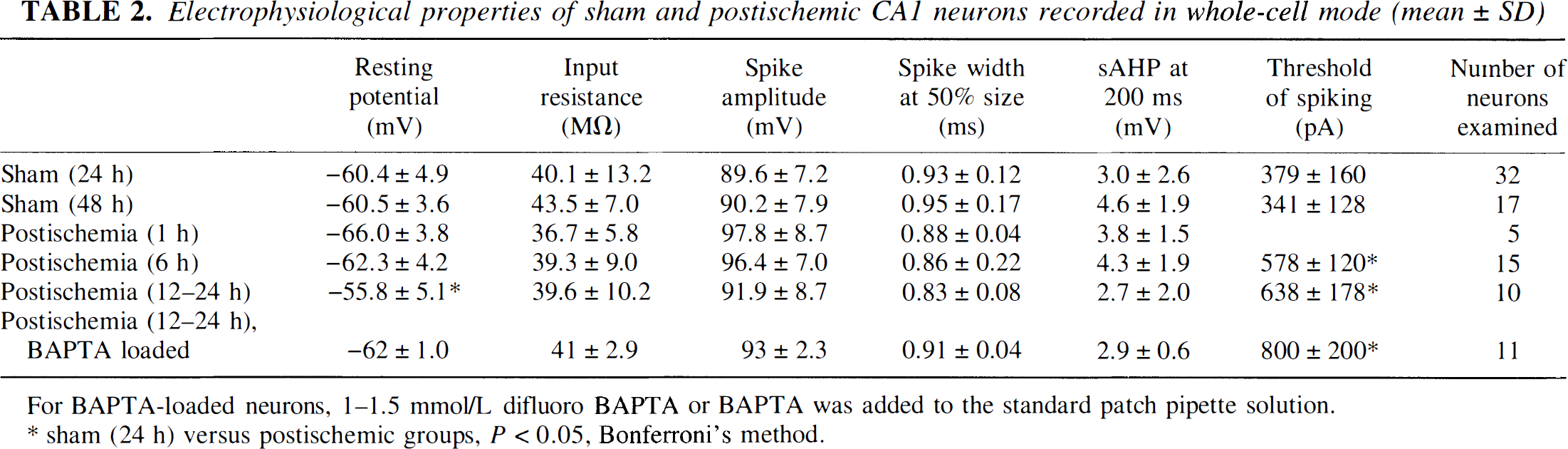
For BAPTA-loaded neurons, 1−1.5 mmol/L difluoro BAPTA or BAPTA was added to the standard patch pipette solution.
sham (24 h) versus postischemic groups, P < 0.05, Bonferroni's method.
Individual sham neurons recorded in whole-cell mode showed graded synaptic responses after stimulation of Schaffer collateral afferents. In current-clamp recordings, these synaptic responses consisted of a fast EPSP after by a monophasic inhibitory postsynaptic potential (IPSP) (Fig. 6, a1). When neurons were voltage-clamped at potentials near −80 mV, which is close to the reversal potentials for Cl−- and K+-mediated conductances, Schaffer collateral stimulation induced an inward, excitatory postsynaptic current (EPSC) (Fig. 6, a2). At the negative holding potential, paired stimuli of Schaffer collateral afferents induced an enhancement of the EPSC by 55 ± 19% (n = 25) and 56 ± 17% (n = 14) in slices prepared 24 and 48 hours after surgery, respectively.
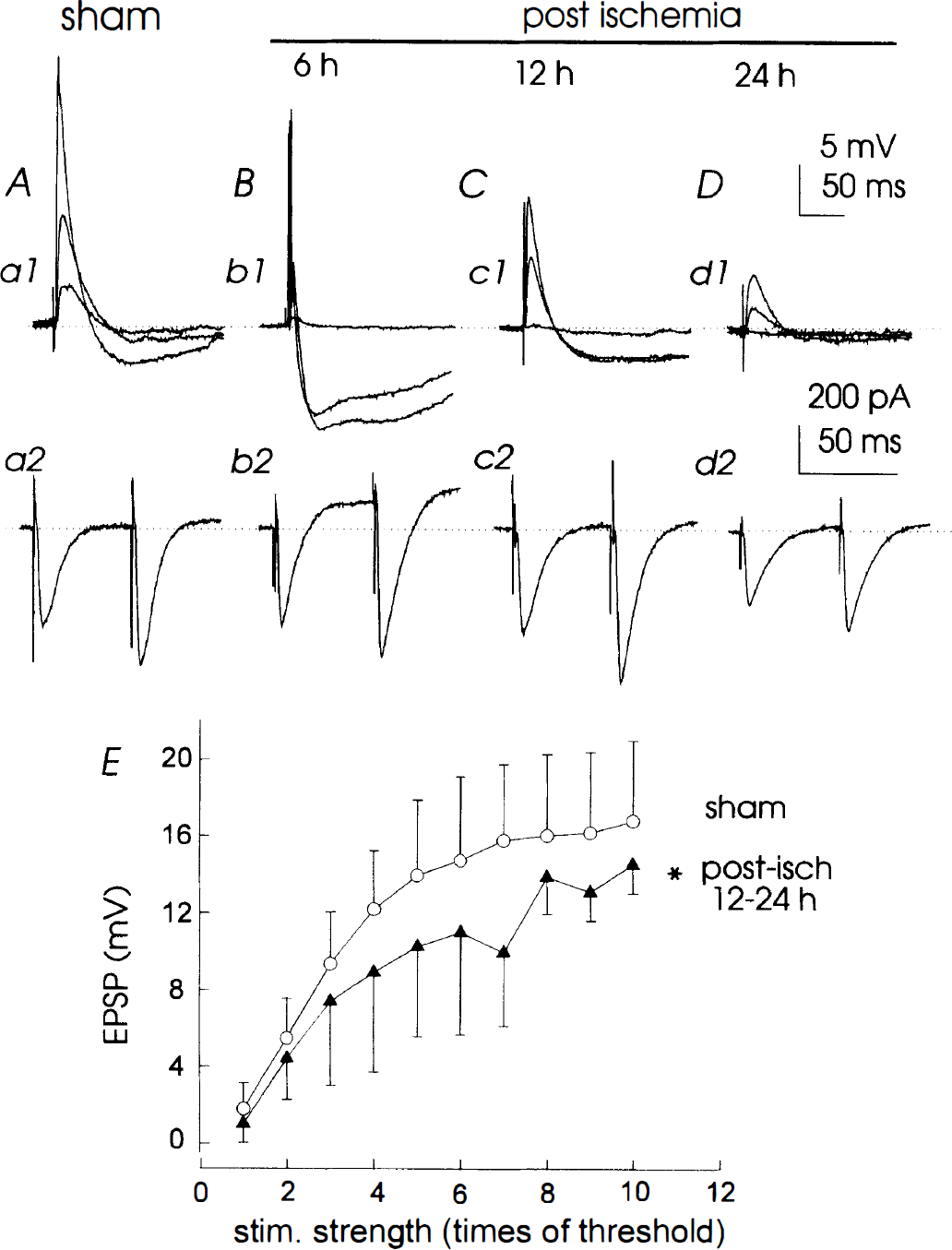
Decreased synaptic responses in postischemic CA1 neurons recorded in the whole-cell mode. Synaptic events were collected from four individual CA1 neurons in slices prepared at 24 hours after sham surgery
Postischemic CA1 neurons. We had no difficulty in performing whole-cell recordings on CA1 neurons from slices prepared 6 hours after reperfusion. CA1 neurons recorded from these slices exhibited basic membrane properties that were similar to those of sham controls (Table 2). However, large depolarizing current pulses were required to evoke action potentials in these neurons, suggesting that an elevated spiking threshold exists in postischemia neurons, even after 6 hours of reperfusion (Table 2). At the single-cell level, individual CA1 neurons clearly showed synaptic events after stimulation of Schaffer collateral afferents, and the EPSC were enhanced by 48 ± 15% (n = 6) after the paired afferent stimulation at negative holding potentials. Interestingly, stimulating Schaffer collateral afferents often induced large, biphasic IPSP that became dominant at the resting potentials near −60 mV, thus masking the depolarizing EPSP (Fig. 6, b1). To reveal the potential excitatory synaptic events, neurons were voltage-clamped at −80 mV to decrease the driving force for the Cl− and K+ conductances. EPSC of similar magnitude to sham were revealed after afferent stimulation at the negative holding potential (Fig. 6 b2). These observations suggest that the large IPSP we observed in these neurons results from enhanced synaptic inhibition, rather than diminished excitatory synaptic events.
Whole-cell recordings on CA1 neurons were difficult to achieve in slices prepared 12 or 24 hours after reperfusion, mainly because of the lack of a tight seal between patch pipette tip and the cell membrane. This may result from ischemia-induced alterations in membrane structures of the vulnerable CA1 neurons (Kirino et al., 1992). To circumvent this, we have used different patch pipette glasses or patch pipette filling solutions, including the solution containing 1 to 1.5 mmol/L potassium salt of BAPTA or 4,4′-difluoro BAPTA. However, no consistent improvement was noted when doing whole-cell recordings on these neurons. We listed separately the whole-cell measurements in neurons dialyzed with the standard or BAPTA-containing patch pipette solutions (see Table 2). Although no major difference was observed between these two solutions, a small hyperpolarization of the membrane potential by 5 mV in BAPTA-dialyzed neurons was noted. In these limited whole-cell recordings, CA1 neurons 12 to 24 hours after ischemia displayed basic membrane properties comparable with those of sham controls (Table 2). In response to depolarizing current pulses, however, these neurons, dialyzed with either the standard or BAPTA-containing patch pipette solution, displayed tonic discharges with little firing adaptation in comparison with sham controls (Fig. 5, c1, d1). The current threshold for the generation of action potentials was significantly increased to 638 to 800 pA, which is nearly double that of sham controls (Table 2). Furthermore, the synaptic responses, as well as the enhancement after paired stimulation of Schaffer collateral afferents at negative holding potentials (41 ± 12%, n = 12), were significantly smaller in neurons 12 and 24 hours after ischemia than those observed in sham controls (Fig. 6E; Table 2).
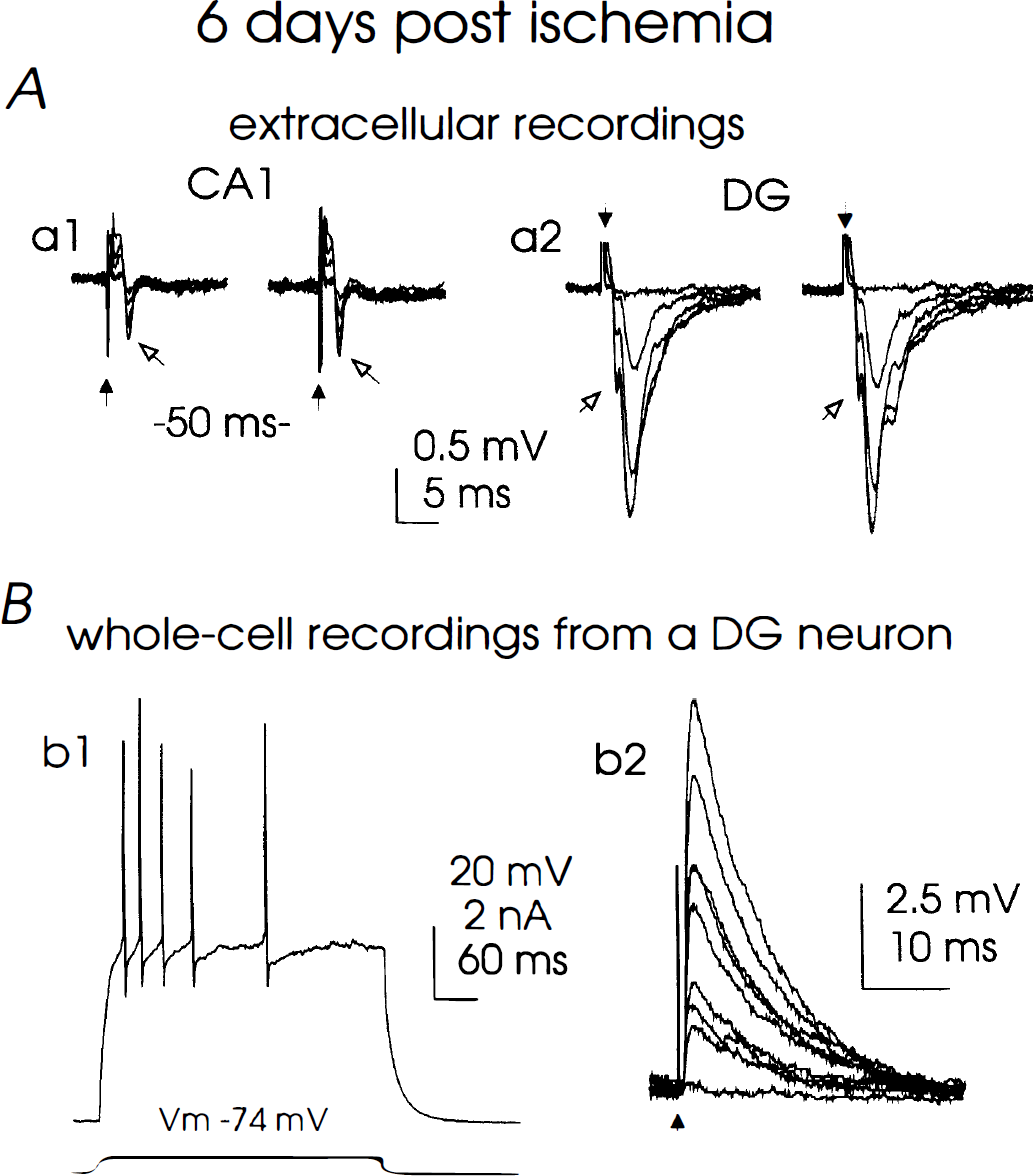
Electrophysiologic responses at 6 to 7 days after ischemia reperfusion.
In contrast to the abolished synaptic responses in the CA1 region, stimulation of the perforant pathway induced synaptic field potentials in the region of the dentate gyrus in the same slices prepared 6 to 7 days after ischemia (Fig. 7A, a2). The maximal dendritic field EPSP in the dentate gyrus was 0.94 ± 0.15 mV, which was significantly smaller than that measured from sham slices (1.82 ± 0.39 mV, n = 8 from 2 rats). We readily achieved whole-cell recordings on dentate gyrus neurons 6 to 7 days after ischemia, using patch pipettes filled with our standard solution. These neurons displayed resting potentials of −75 ± 2 mV, input resistances of 115 ± 3 MΩ, spike amplitudes of 109 ± 5 mV, and slow after-hyperpolarizations of 8.9 ± 0.5 mV at −60 mV (n = 4). At the single-cell level, all dentate gyrus neurons examined displayed repetitive discharges and graded synaptic responses (Fig. 7B, b1, b2), and these electrophysiologic parameters are within the range measured from naive dentate gyrus neurons under the whole-cell mode (Staley et al., 1992; Zhang, unpublished data). Taken together, these results indicate that electrophysiologic activities of dentate gyrus neurons are largely preserved at prolonged times after postischemia reperfusion.
DISCUSSION
The key finding we present in this study is that transient global ischemia promotes alterations in the functionality of hippocampal CA1 neurons at times preceding their degeneration as identified by morphologic criteria. We demonstrate that within 24 hours after ischemia reperfusion, CA1 neurons display a dramatic, time-dependent alteration in synaptic activities. CA1 synaptic responses are largely intact after 6 hours of postischemia reperfusion, become significantly impaired by 12 hours, and continue to deteriorate until and after 24 hours of reperfusion. Furthermore, decreased synaptic responses observed at 12 hours after ischemia differ from those observed 24 hours after ischemia. This is shown by the restoration of synaptically evoked PS after the blockade of GABAA receptors after 12 hours, but not after 24 hours of reperfusion. Additionally, we observed a time-dependent increase in the firing threshold in postischemic CA1 neurons, which is consistent with the idea that both synaptic and intrinsic activities of postischemic CA1 neurons are down-regulated before substantial changes in their morphologic features at the light-microscopic level.
The decrease in intrinsic excitability in postischemic neurons is manifested by an increase in the firing threshold, as the minimum positive current required to generate action potentials through patch pipettes is doubled in postischemic neurons. Since the resting potential, input resistance, and action potential waveform of postischemic neurons do not differ substantially from those of sham controls, the increase in the firing threshold must result from alterations in the magnitude or the kinetics of tetrodotoxin-sensitive Na+ currents. It is well known that the activities of Na+ channels are down-regulated through kinase-dependent modulation (Catterall, 1992), and we propose that a similar negative modulation of Na+ channels may occur in these postischemic neurons. It will be of considerable interest for future experiments to characterize Na+ channel activities and their modulation after ischemia.
We demonstrate that although the magnitudes of CA1 synaptic responses are decreased to a similar extent at 12 and 24 hours after reperfusion, the responses to bicuculline, a selective GABAA receptor antagonist, are different. The blockade of GABAA receptors by bicuculline restored synaptically induced neuronal discharge in some CA1 neurons 12 hours after ischemia, but failed to do so in CA1 neurons 24 hours after the insult. Explanations for these findings are multiple. First, it is likely that the GABAA-mediated synaptic inhibition in the CA1 region is only partially impaired in slices prepared 12 hours after ischemia. Thus, a blockade of functional GABAA receptors boosts the synaptic excitation in these slices. Whereas if the GABAA-mediated synaptic inhibition is severely impaired in slices prepared 24 hours after ischemia, the blockade of dysfunctional GABAA receptors would have little influence on the state of synaptic activity. However, it is equally possible that glutamate-mediated synaptic events decrease similarly during postischemia reperfusion. Thus, because of a substantial loss in the excitatory drive to CA1 neurons at 24 hours after ischemia, the rebound excitation resulting from GABAA blockade would not be evident, even if some functional GABAA receptors do remain. Although we have not investigated these possibilities in the current experiments, it is suspected that several mechanisms coexist in the postischemic state. Nevertheless, the current results are consistent with previous studies, which show that GABA-ergic neurons in the CA1 region are more resistant to ischemic insults than pyramidal neurons (Johansen et al., 1989; Nitsch et al., 1989; Schandler et al., 1988), and support the recent study (Schwartz et al., 1995) that potentiation of GABAA-mediated inhibition by diazepam early during reperfusion is protective in a similar global ischemia model.
Postischemic CA1 neurons in brain slices displayed decreased synaptic responses after stimulation of Schaffer collateral afferents. The decrease in CA1 synaptic events was substantial when examined at 12 hours after ischemia, and the synaptic responses continued to deteriorate throughout our recording times in the CA1 region. This differs from previous studies using a similar four-vessel occlusion model, where CA1 synaptic activity was found intact at 24 hours after reperfusion (Petito and Pulsinelli, 1984; Jensen et al., 1991). The reasons for the discrepancy between our results and these reports are not clear. We conducted the four vessel occlusion model a modified method as per Pulsinelli and Brierley (1979). Morphologic and electrophysiologic assessments from isolated slices were done only if EEG activities of these animals were completely abolished during ischemia, and the blood gas parameters were maintained within restricted ranges (see Methods). In our hands, the insult we conducted reproducibly produced a neuronal loss of 95% or more in the CA1 region by day 7 after ischemia. It is speculated that severe ischemic insults conducted in the current experiments promotes the early appearance of postischemia dysfunction, and that differences in the severity of ischemic injury may influence the time frame of neuronal degeneration, as has been suggested by Du and associates (1996) in a focal ischemic model.
In the current experiments, we examine the electrophysiologic properties of postischemic CA1 neurons in brain slices as a function of time. Brain slices were chosen for our investigations because local synapses are largely preserved in this isolated preparation, and electrophysiologic recordings in brain slices are conducted without the complications that often accompany in vivo experimental conditions such as anesthesia and stress. Synaptic field potentials in brain slices have been widely used to assess the efficiency of synaptic transmission in hippocampal neurons (Lynch and Schubert, 1980; Schwartzkroin, 1981). We propose that delayed neuronal death must be associated with or preceded by functional alternations of the CA1 neurons, or related local structures that influence these neurons. The current observations (i.e., that altered electrophysiologic properties are observed in CA1 neurons of isolated brain slices starting at 12 hours or less after reperfusion) are consistent with this hypothesis. However, the use of brain slices has limitations, and one main drawback is the lack of synaptic innervation to the hippocampus from extrahippocampal structures. These structures not only undergo postischemic degeneration themselves (Rod and Auer, 1992) but also may participate in the genesis of the delayed neuronal death that occurs in the CA1 neurons (Buchan et al., 1991a). Thus, it is conceivable that the electrophysiologic activities of postischemic CA1 neurons are far more complex in vivo than in vitro (Buzsaki et al., 1989; Chang et al., 1989; Furukawa et al., 1990; Miyazaki et al., 1993; Suzuki et al., 1983; Xu and Pulsinelli, 1996). The current results demonstrate changes observed in our slice preparation, primarily involving CA1 synapses and their local structures.
Another complication regarding electrophysiologic studies on brain slices is that the observed changes may include factors induced by slicing procedures. Although the sham and postischemic slices were prepared similarly in the current experiments, it is possible that postischemic brain tissues are more susceptible than sham to the damage or deterioration resulting from slicing procedures and incubation in vitro, thereby leading to altered electrophysiologic responses. However, because the electrophysiologic activities observed in dentate gyrus neurons were largely preserved in slices prepared from animals 6 to 7 days after reperfusion, and the activities of CA1 neurons in the same slices were completely abolished, we suggest that decreased synaptic activities in postischemic CA1 neurons result predominantly from ischemic insults rather than damage incurred by slicing procedures.
We show here that postischemic CA1 neurons display hypoactivity rather than hyperactivity in glutamate-mediated synaptic transmission, at least in the Schaffer collateral–CA1 synapses examined in our isolated brain slices. Although we have not identified the presynaptic or postsynaptic components of this alteration, our description of decreased activities is consistent with a previous molecular study that demonstrates decreases after ischemia in the expression levels for genes encoding the AMPA/kainate subtype of glutamate receptors (Pellegrini-Giampietro et al., 1992). Since these receptors represent the major participants involved in fast glutamatergic synaptic transmission, such a decrease in mRNA prevalence could lead to fewer receptors, and therefore less glutamate activity, as we demonstrate in this study. Although the pathophysiologic significance of the decreased synaptic activities early during reperfusion has not been demonstrated, we speculate that any alteration of neuronal function from normal may participate in or represent some form of the pathologic process underlying the delayed neuronal death.
It has been recognized that the survival of developing neurons in vitro requires neurotrophic factors as major supporting elements (Purves et al., 1985), and this neurotrophic support is not promoted unless some ongoing neuronal activities are present (Meyer-Franke et al., 1995). In mammalian CNS preparations, neuronal activities and neurotrophic factors interact reciprocally such that external applications of neurotrophins, such as BDNF and NT-3, produce persistent potentiation of glutamate transmission at Schaffer collateral–CA1 synapses in adult rat hippocampal slices (Kang and Schuman, 1995). Furthermore, the release of nerve growth factor is positively regulated in hippocampal slices and in cultured hippocampal neurons in an activity-dependent manner (Blöchl and Thoenen, 1995). Considering that the delayed neuronal death after transient ischemia is at least partly mediated by apoptotic mechanisms (Nitatori et al., 1994), the early and persistent decreases in synaptic activities in vulnerable postischemic CA1 neurons may have significant impact that leads toward the apoptotic process, perhaps through disruption of neurotrophic support. It remains to be tested whether maintenance of synaptic activities to a certain level benefits the survival of CA1 neurons, particularly after prolonged postischemic reperfusion.