
Abstract
MicroRNA (miRNA), miR-181a, is enriched in the brain, and inhibition of miR-181a reduced astrocyte death
INTRODUCTION
Several clinical situations such as cardiac arrest and resuscitation or severe hypotension can lead to transient global cerebral ischemia. Global ischemia causes selective, delayed death of pyramidal neurons in the hippocampal cornus ammonis (CA)1 while sparing the neighboring dentate gyrus in rodents 1 and humans, resulting in persistent severe memory deficits.2, 3 However, despite increasing knowledge of the pathologic mechanisms of global ischemia, the only known clinically effective treatment is hypothermia.4, 5 Neuronal death in CA1 is not histologically detectable until 3 to 4 days after global/forebrain ischemia, and this significant delay between injury and the onset of neuronal death holds promise for the development of future therapies, including ones targeting apoptosis.6, 7, 8
MicroRNAs (miRNAs) are small regulatory noncoding RNAs ∼22 bp in length that modulate protein synthesis by post-transcriptional silencing of gene expression via the recognition of specific sequences in target messenger RNAs (mRNAs). Many miRNAs are highly expressed in the adult nervous system in a regulated manner, and pathologic conditions including focal and global ischemia lead to altered regulation of brain miRNAs.9, 10, 11 These reports suggested that miRNAs could have a critical role in molecular signaling associated with ischemic pathologic events. Indeed, several laboratories have demonstrated that manipulation of miRNAs can attenuate infarct volume in a focal ischemia model.12, 13, 14 However, in contrast to focal ischemia, it still remains unknown whether modulation of miRNAs can protect against global ischemia.
miR-181a, a miRNA that is highly conserved across most vertebrates, is enriched in the brain15, 16 and increases during maturation of hippocampal neurons. 17 The overexpression of miR-181a induces drug addiction-related synaptic changes in the hippocampus 17 and increases astrocyte death because of glucose deprivation. 18 miR-181a is upregulated in the infarct core and downregulated in the penumbra after focal ischemia14, 17 but it has not yet been studied in forebrain ischemia. In the present study, we test the effect of reducing miR-181a on forebrain ischemia, and, because of the importance of apoptosis in this setting, characterize the effects on levels of Bcl-2, a target of miR-181a. We also examined the effect of miR-181a in N2a cells and primary neurons subjected to stress.
MATERIALS AND METHODS
Cell Culture and Transfection
Mouse neuroblastoma (N2a) cells (a kind gift from Drs Kurt Lucin and Tony Wyss-Coray at Stanford University) were grown in high-glucose Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA, USA) supplemented with 8% fetal bovine serum (Hyclone, Logan, UT, USA) and 1% antibiotics (50 U/mL penicillin and 50
Primary mouse neuronal cultures were prepared from 16-day-old embryos as previously described
19
in accordance with a protocol approved by the Stanford University Animal Care and Use Committee and after the NIH
Injury Paradigms
Both N2a cells and primary neurons were subjected to injury 24 hours after transfection.
N2a cells were exposed to 900 μ
Quantitation of Cell Death
The viability of N2a cells was quantified by measuring the concentration of intracellular lactate dehydrogenase (with LDH kit from Sigma) released from dead cells into the media. 20 The results were expressed as the percentage of lactate dehydrogenase release compared with lactate dehydrogenase release after freeze thaw to disrupt all cells.
The viability of primary neurons was assessed by cell counting after Hoechst 33342 (5
Imaging-Activated Caspases
N2a cells were stained with Magic Red (Immunochemistry, Bloomington, MN, USA) to identify cells with activated caspases 3 and 7 and counterstained with Hoechst 33342 (5
Animal Groups
Male Sprague–Dawley rats (Charles River, Wilmington, MA, USA) weighing 280 to 320 g were divided into the following groups: sham (
Transfection of Rat Hippocampus In Vivo
Rats were anesthetized with isoflurane and placed in a stereotaxic frame with a rat head holder. Three picomoles per gram of miR-181a antagomir or mismatch miR-81a antagomir (Thermo Scientific Dharmacon), mixed with the cationic lipid DOTAP 1:3 (Roche Applied Science, San Francisco, CA, USA), were infused stereotactically just outside the left hippocampus (from bregma −3.8 mm, M-L 2.0 mm, deep 2.5 mm) at 1
Transient Forebrain Ischemia
Forebrain ischemia was induced 24 hours after transfection
22
according to an animal protocol approved by the Stanford University Animal Care and Use Committee. Briefly, hypotension (mean blood pressure <40 mm Hg) was induced by removing blood into heparinized sterile tubing with continuous femoral arterial blood pressure monitoring (Puritan–Bennett Corporation, Wilmington, MA, USA). Both carotid arteries were clamped for 10 minutes, then unclamped and the shed blood reinfused. Rectal temperature was 37±0.5 °C controlled by a homeothermic blanket (Harvard Apparatus, Holliston, MA, USA). Respiratory rate, oxygen saturation, and heart rate were monitored with a small animal oximeter (STARR Life Sciences, Allison Park, PA, USA). After forebrain ischemia, rats were killed at 5 or 24 hours after ischemia for protein and RNA isolation. Control rats injected with antagomir or mismatch but not subjected to ischemia were killed 1 and 3 days after injection to assess protein and miRNA levels. For assessment of CA1 neuronal survival by cresyl violet staining or assessment of protein by immunoblotting, rats were killed at 7 days after ischemia under deep anesthesia with isoflurane. Brains were collected after perfusion with cold saline for protein or RNA, or saline followed by ice-cold 4% phosphate-buffered paraformaldehyde for cresyl violet staining. After 48 hours after fixation in 4% paraformaldehyde, 50
Assessment of CA1 Hippocampal Injury
Coronal sections were mounted on gelatin-coated slides and then incubated in 1% cresyl violet acetate (Sigma) for 5 minutes followed by dehydration. The density of cresyl violet staining was determined in a fixed area of CA1 on each micrograph using the histogram function of Adobe Photoshop CS3 and normalized to sham by a researcher masked to treatment, as previously described. 21
Immunoblotting
Immunoblotting was performed as previously described.
22
Homogenates prepared from either cell cultures or subdissected CA1 and dentate gyrus regions of the hippocampus from brain were studied. Briefly, 20 to 40
Real-Time Quantitative PCR
Total RNA was extracted with TRIzol reagent (Invitrogen), and the concentration and purity of the RNA samples were quantified by absorbance at 260 and 280 nm. Reverse transcription of miRNA was performed using the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA, USA). Equal amounts of RNA (200 ng) were reverse transcribed with 1.3 m
Statistical Analysis
All cell culture data represent at least three independent experiments. All data reported are means±s.e.m. Statistical analysis was performed using
RESULTS
Altering miR-181a Levels Alters Bcl-2 Levels and Apoptotic Cell Death of N2a cells
To investigate the effect of altering miR-181a levels on N2a cells, cells were transfected with miR-181a mimic, miR-181a inhibitor, or control. Treatment with mimic increased miR-181a levels 4,700-fold, whereas a decrease of 88% was seen with the inhibitor (Figure 1A). As we previously found that Bcl-2 was a target of miR-181a in astrocytes, we therefore determined whether changing levels of miR-181a would affect Bcl-2 levels in N2a cells. miR-181a inhibitor treatment significantly increased Bcl-2 protein levels, whereas mimic-treated cells had decreased levels relative to control (Figure 1B).
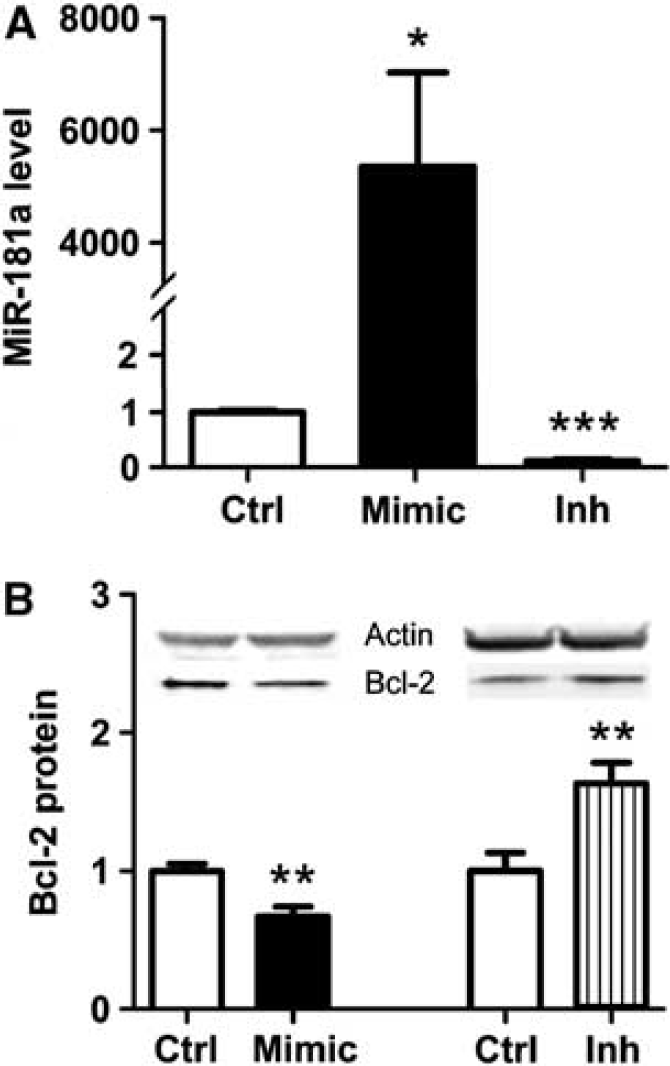
The effect of mimic and inhibitor on miR-181a level and Bcl-2 protein expression in N2a cells. (
N2a cells were next exposed to combined serum deprivation and oxidative stress for 24 hours to induce apoptosis. Overexpression of miR-181a significantly increased cell death, whereas inhibitor effectively reduced cell death (Figure 2A). There was no significant difference in cell death between injury control without (data not shown) vs with treatment with transfection control (Trans). To confirm that cell death was apoptotic, we stained the N2a cells with Magic Red to detect active caspases 3 and 7. Increasing miR-181a by treatment with mimic was associated with many more cells showing activation of caspases 3 and 7, whereas reduction of miR-181a by inhibitor treatment reduced the number of cells activating these enzymes (Figures 2B and C).
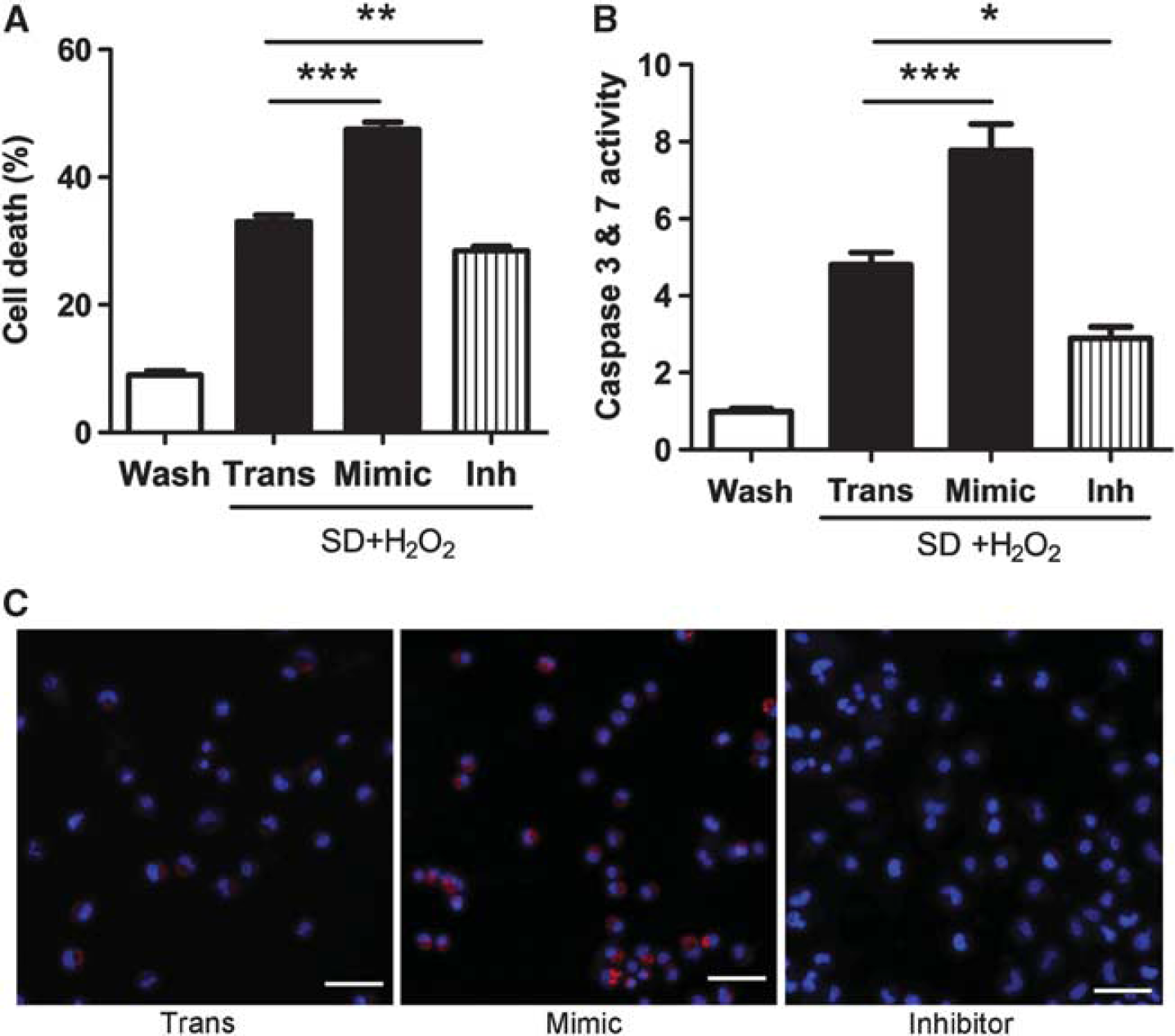
Decreasing miR-181a levels reduces serum deprivation and oxidative stress-induced N2a cell death and caspases 3 and 7 activation. (
Effect of Altering miR-181a Levels in Primary Neurons
We compared the expression of miR-181a in primary neurons and N2a cells by real-time quantitative PCR. There was no difference in threshold cycle for U6 between the two cell types, but the threshold cycle for miR-181a was significantly lower in primary neurons (20.09±0.03 vs 25.92±0.28 for N2a cells;
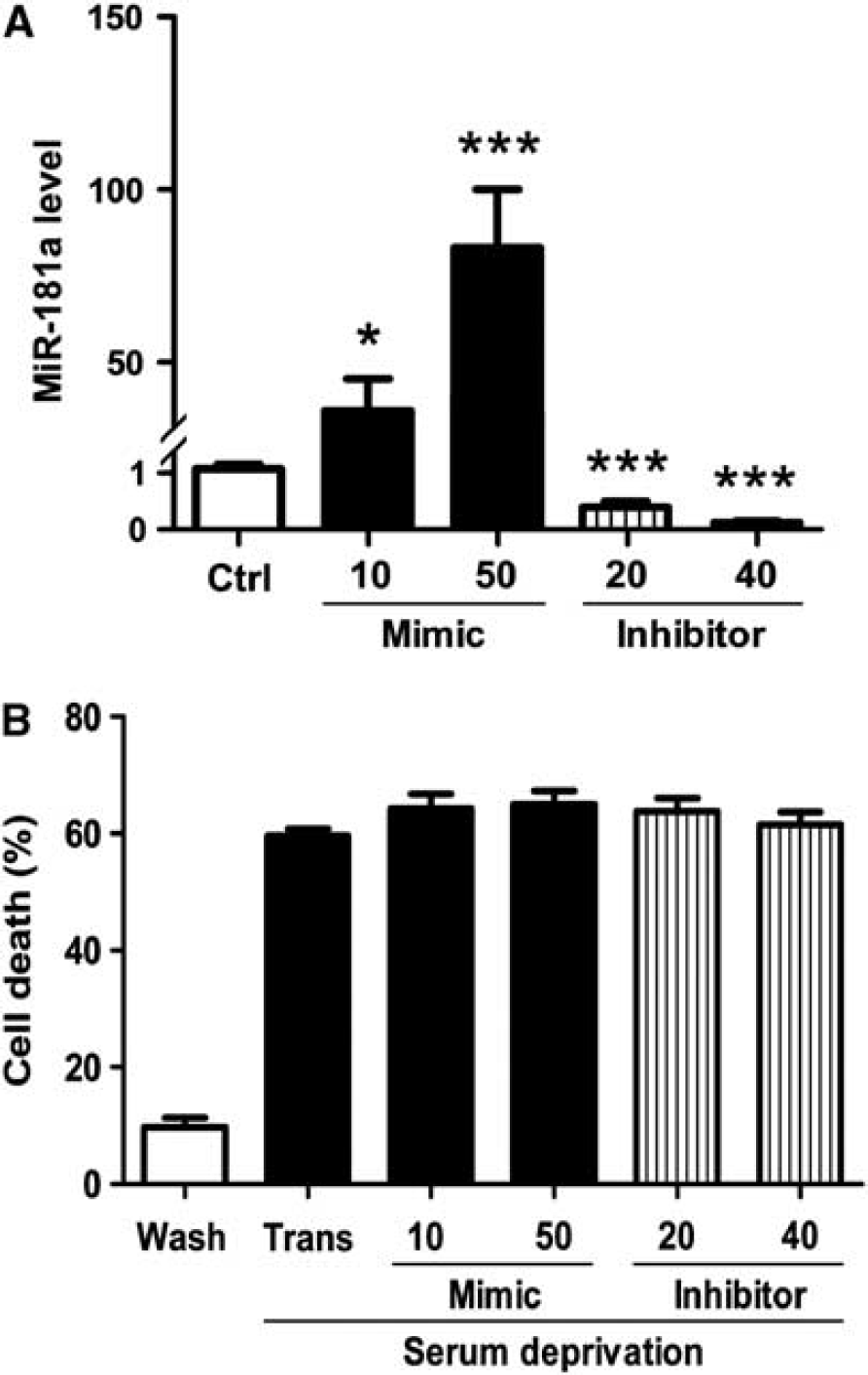
Altering miR-181a levels in primary neurons does not alter serum deprivation cell death. (
Forebrain Ischemia Changes miR-181a and Bcl-2 Protein Levels in CA1 and Dentate Gyrus in the Hippocampus
CA1 is the subregion of the hippocampus most susceptible to ischemia. We assessed levels of miR-181a 5 and 24 hours after 10 minutes of forebrain ischemia. In CA1, miR-181a significantly increased 24 hours after ischemia (Figure 4A), whereas Bcl-2 protein decreased (Figure 4B). In contrast, in the dentate gyrus miR-181a tended to decrease at 5 hours, whereas Bcl-2 protein was increased at both 5 and 24 hours (Table 1).
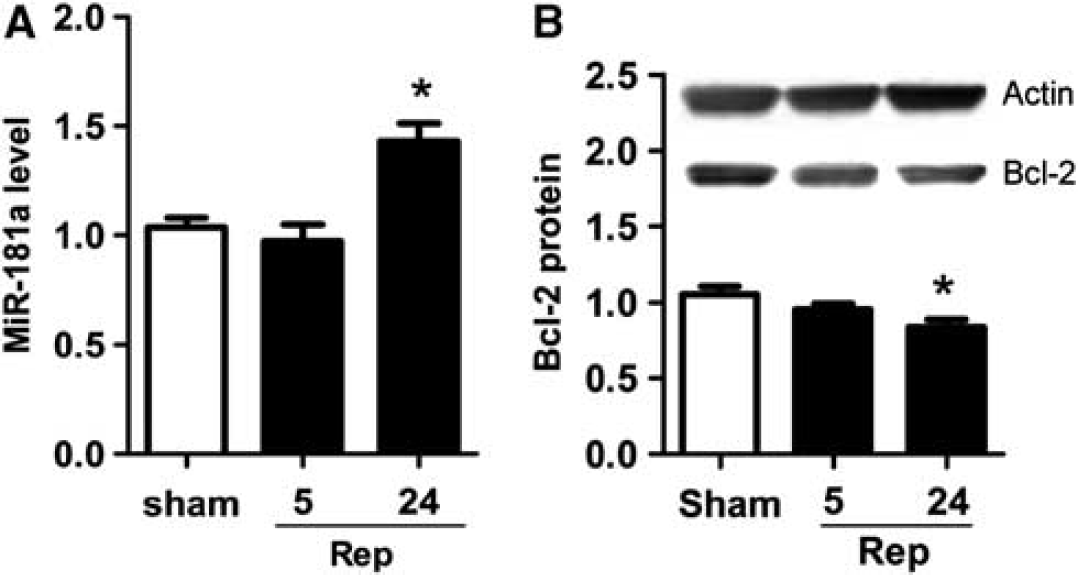
Temporal change of miR-181a and Bcl-2 protein in the CA1 region of hippocampus after forebrain ischemia. (
Levels of miR-181a and Bcl-2 protein in the dentate gyrus region of hippocampus after forebrain ischemia

Values are mean±s.e.m.
∗
∗∗
Reducing Levels of miR-181a Reduces Forebrain Ischemic Injury
Real-time quantitative PCR was performed 1 and 3 days after injection to determine the extent to which miR-181a levels changed after transfection with antagomir by stereotactic injection. miR-181a antagomir, but not mismatched antagomir, significantly reduced miR-181a expression 1 and 3 days after a single treatment (Figure 5A). Bcl-2 protein levels also changed with a significant increase on day 1 and a trend to increase on day 3 (Figures 5B and 5C). Therefore, we performed forebrain ischemia on rats 1 day after stereotactic infusion of antagomir or control. Cresyl violet staining performed 7 days after forebrain ischemia revealed that when brains were transfected with miR-181a antagomir CA1, neuronal cell death was significantly reduced compared with rats treated with mismatch miR-181a antagomir (Figures 5D and 5E).
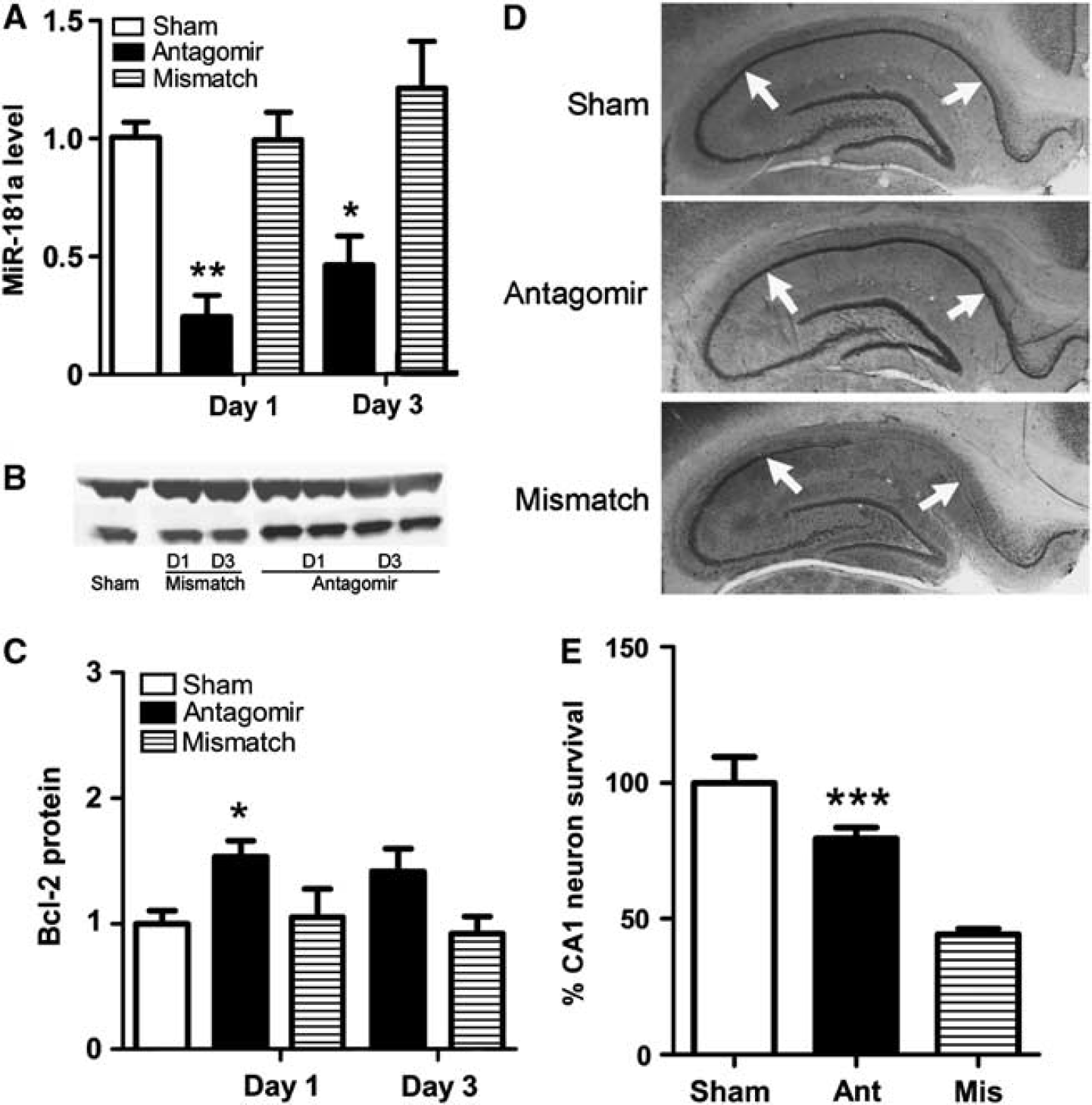
Pretreatment with miR-181a antagomir increases CA1 neuronal survival after forebrain ischemia. (
The Effect of miR-181a Antagomir on GLT-1 in CA1 After Forebrain Ischemia
As we previously found that astrocyte impairment was a critical determinant of the extent of subsequent neuronal cell death in forebrain ischemia, 22 we hypothesized that miR-181 antagomir might reduce astrocyte impairment. Earlier studies showed that reducing miR-181a levels in primary astrocyte cultures could reduce oxidative stress, 18 and oxidative stress contributes to astrocyte impairment and reduced GLT-1 levels after forebrain ischemia. We therefore determined the level of GLT-1 in the CA1 subfield by immunoblot analysis. GLT-1 expression significantly decreased in CA1 24 hours after forebrain ischemia compared with sham; however, treatment with miR-181a antagomir significantly inhibited this decrease of GLT-1 compared with vehicle or mismatch control antagomir (Figure 6).
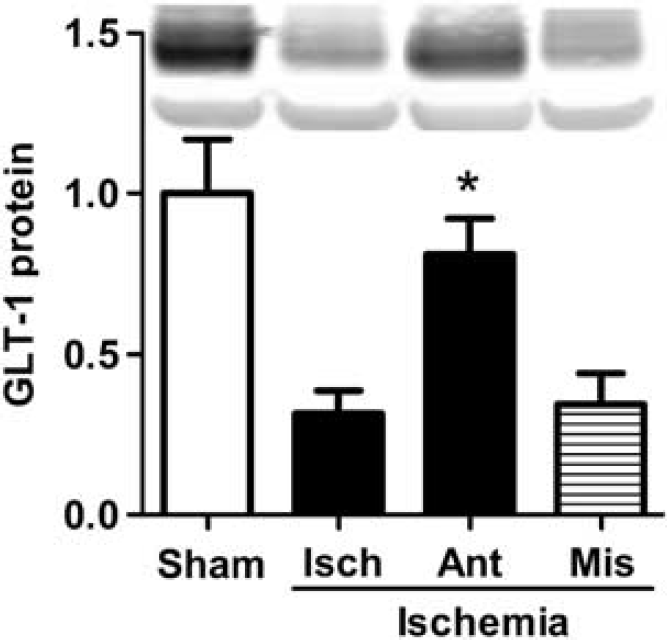
Pretreatment with miR-181a antagomir preserves GLT-1 expression after forebrain ischemia. GLT-1 expression 24 hours after reperfusion in the CA1 subfield of rats treated with miR-181a antagomir, mismatch miR-181a antagomir, vehicle (Isch), and sham rats, which were not subjected to ischemia. Inset shows representative western blot.
DISCUSSION
Pretreatment with miR-181a antagomir reduces loss of CA1 pyramidal neurons 7 days after forebrain ischemia. This adds forebrain ischemia to the settings in which reducing miR-181 provides neuroprotection. Treatment with antagomir inhibited the changes seen in untreated animals, reducing miR-181a, increasing Bcl-2 protein levels, and preserving neurons in CA1. Although pretreatment was tested in this study, future studies of the effect of changing miRNA levels after the onset of ischemia will be an essential step in further demonstrating the likely relevance for clinical treatment.
Overexpression of Bcl-2, the prototype anti-apoptotic member of the eponymous family of apoptosis regulatory proteins, was previously shown to be protective against both focal and global cerebral ischemia
Although we demonstrate changes in one target of miR-181a, a limitation of our study is that there are likely to be additional targets of miR-181a that may have a role in the protection we observe, because miRNAs target multiple mRNAs. Our prior work showed that miR-181a can directly target Mcl-1 and Grp78/BIP in some cells,14, 18 and a recent report showed that X-linked inhibitor of apoptosis is also a direct target, and that several antioxidant enzymes are likely targets. 28 These targets, which are all pro-survival proteins and can reduce oxidative stress, could also have a role here. We think this is relevant because our earlier studies demonstrated a link between oxidative stress in astrocytes, reduction in GLT-1, and CA1 neuronal cell death.21, 22 miR-181 was also shown to sensitize glioblastoma cells to apoptosis by reducing Bcl-2. 29 Future studies are needed to explore additional potential targets in this setting.
One interesting observation is the difference in effect of miR-181a antagomir in different cell types. Reducing miR-181a increased Bcl-2 in N2A cells and increased Bcl-2, Grp78, and Mcl-1 in primary astrocytes while increasing survival of both these cell types. It failed to significantly change levels of Bcl-2 in primary neurons, and it did not change outcome from serum deprivation-induced neuronal cell death. This observation is consistent with an increasing body of literature that suggests that individual miRNAs may have modest effects on their target mRNAs, and that often several miRNAs are required for larger, or in some cases, detectable alteration of target protein levels. In prostate cancer cells, miR-181a acts cooperatively with two other miRNAs to regulate Grp78 protein levels, 30 in contrast to the response of astrocytes in which increasing miR-181a alone effectively downregulates Grp78.14, 30 Further, cell lines are in general dependent on inducing anti-apoptotic pathways for survival; hence, the N2A cells may be more sensitive to the ability of miR-181 to reduce several anti-apoptotic proteins than are primary, postmitotic neurons. Thus, the efficiency of regulation of a target protein depends on the cell type and likely the physiologic setting; hence, the difference observed between N2A cells and primary neurons may not be surprising.
Total hippocampal miR-181a was reported to be upregulated 30 minutes and 24 hours after 20 minutes of global ischemia, 31 and hippocampal Bcl-2 protein levels were previously found to be decreased 24 hours after 10 minutes of global ischemia. 32 Although these studies evaluated changes in whole hippocampus, we dissected out the selectively vulnerable CA1 region so as not to average changes in the vulnerable region with changes in the other hippocampal regions that survive. We found that miR-181a increased in CA1 but tended to decrease in dentate gyrus (Table 1), whereas Bcl-2 protein levels decreased in CA1 but increased in dentate gyrus. These results are consistent with the pattern of neuronal cell death in CA1 and survival in dentate gyrus.
The significant protection observed here against forebrain ischemia was associated with increased Bcl-2 protein levels and preservation of the astrocyte glutamate transporter GLT-1. We previously reported that inhibition of miR-181a in primary astrocytes increased Bcl-2 protein and reduced oxidative stress during glucose deprivation. 18 A close association was seen between oxidative stress, the early loss of astrocyte GLT-1, and delayed CA1 neuronal death in forebrain ischemia.21, 22 Thus, the preservation of GLT-1 observed here is consistent with protection of astrocyte function contributing to neuronal protection. Astrocytic glutamate uptake prevents or limits excitotoxic neuronal injury.21, 22, 33, 34, 35 In global and forebrain ischemia, reduced astrocytic glutamate uptake precedes neuronal loss. Preventing this astrocyte dysfunction with metabotropic glutamate receptor agonists 35 or by overexpressing protective proteins selectively in astrocytes21, 22 reduces neuron cell death. In this study, significant decreases in GLT-1 protein were demonstrated 24 hours after ischemia, consistent with other studies in gerbils and rats,22, 36 and antagomir treatment reduced this loss.
Despite the observed changes in GLT-1, miR-181a does not directly target GLT-1. Immunoblotting did not show an increase of GLT-1 protein in rats pretreated with miR-181a antagomir (data not shown), and no potential binding sites were identified in the 3'-untranslated region of GLT-1 mRNA for miR-181a using the miR-Target analysis database (http://www.targetscan.org, http://www.microRNA.org). Thus, the protection is likely indirect secondary to reduced oxidative stress in astrocytes. Reduced oxidative stress may reflect increased levels of Bcl-2, Grp78, and Mcl-114, 18 and possibly glutathione peroxidases 1 and 4, and peroxiredoxin 2. 28
Astrocytes support neurons and other surrounding cells through multiple functions including regulation of blood flow, provision of substrates, metabolic and ionic homeostasis, and antioxidant defense.33, 37, 38 However, after ischemia or other central nervous system insults, astrocytes become reactive and may lose some supportive functions, thereby contributing to neuronal death. 39 Ischemic injury induces activation of astrocytes, early loss of GLT-1 in the CA1 region, with recovery over days after forebrain ischemia. Findings in forebrain ischemia are consistent with reports using the four vessel occlusion model of global ischemia.40, 41 Thus, beneficial effects of miR-181a on astrocytes may lead to reduced or modulated activation and better preserved protective function. Effects of miR-181a antagomir in multiple cell types are probably important to the final extent of protection observed.
SUMMARY
Stereotactic delivery of miR-181a antagomir just outside the hippocampus reduced neuronal cell death in rat forebrain ischemia, associated with increased Bcl-2 and preserved GLT-1 levels, consistent with protection that includes preserving astrocyte function.
Footnotes
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
The authors thank Robin White for assistance with microscopy, Yibing Ouyang for help preparing figures, and William Magruder for assistance with preparing the manuscript.