
Abstract
Mints (munc18-interacting proteins) are novel multimodular adapter proteins in membrane transport and organization. Mint1, a neuronal isoform, is involved in synaptic vesicle exocytosis. Its potential effects on development of ischemic damage to neurons have not yet been evaluated. The authors examined changes in mint1 and other synaptic proteins by immunohistochemistry after transient global ischemia in mouse hippocampus. In sham-ischemic mice, immunoreactivity for mint1 was rich in fibers projecting from the entorhinal cortex to the hippocampus and in the mossy fibers linking the granule cells of the dentate gyrus to CA3 pyramidal neurons. Munc18-1, a binding partner of mint1, was distributed uniformly throughout the hippocampus, and synaptophysin 2, a synaptic vesicle protein, was localized mainly in mossy fibers. After transient global ischemia, mint1 immunoreactivity in mossy fibers was dramatically decreased at 1 day of reperfusion but actually showed enhancement at 3 days. However, munc18-1 and synaptophysin 2 were substantially expressed in the same region throughout the reperfusion period. These findings suggest that mint1 participates in neuronal transmission along the excitatory pathway linking the entorhinal cortex to CA3 in the hippocampus. Because mint1 was transiently decreased in the mossy fiber projection after ischemia, functional impairment of neuronal transmission in the projection from the dentate gyrus to CA3 pyramidal neurons might be involved in delayed neuronal death.
Excessive synaptic transmission has been implicated in delayed neuronal death after brain ischemia (Meldrum, 1990; Choi and Rothman, 1990). At the synapse, exocytosis of synaptic vesicles releases neurotransmitters and constitutes the first step in synaptic transmission. Several proteins that function in synaptic vesicle exocytosis have been identified (Südhof, 1995); however, the effects of ischemia on these proteins are not completely understood.
Transient global ischemia in rodents is known to induce histologic damage restricted to CA1 pyramidal neurons in the hippocampus (Kirino, 1982). Presynaptic boutons in CA1, however, have been reported to be preserved despite development of delayed neuronal death in gerbils (Johansen et al., 1984; Kirino et al., 1990). Several reports also have described hippocampal immunohistochemical profiles of synaptic vesicle proteins, such as synapsins and synaptophysins, during delayed neuronal death that indicate preservation of these proteins (Kitagawa et al., 1992; Miyazawa et al., 1993). Another report described a transient decrease in immunoreactivity of a synaptic vesicle protein during an early stage of neuronal death in CA1 (Miyazawa et al., 1995). These studies suggest that synaptic vesicle proteins are relatively resistant to ischemia, but the role of these proteins in pathogenesis of ischemic damage has remained unclear.
In addition to the above synaptic vesicle proteins, a number of additional synaptic proteins have been reported to regulate neurotransmitter release. A cascade of protein–protein interactions in presynaptic terminals leads to the final Ca2+-triggered exocytosis at the active zone. Among these proteins, munc18-1 is closely related to the C. elegans unc18 gene, which is essential for acetylcholine release in worms (Hosono et al., 1992; Hata et al., 1993). The binding reactions of munc18-1 and its homology to C. elegans and yeast proteins suggest a role for munc18-1 in synaptic vesicle exocytosis, but its exact function is unclear (Südhof, 1995). Recently, the authors identified a novel protein family, mint as munc18-interacting proteins (Okamoto and Südhof, 1997, 1998). The discovery of mints led to the hypothesis that mints could function to couple syntaxin to other proteins in vesicular membrane traffic (Okamoto and Südhof, 1997).
The mint family consists of three members termed mint1, 2, and 3, or alternatively “X11 proteins” (Duclos et al., 1993). Mint family members are multimodular adapter proteins with a unique domain structure composed of an N-terminal sequence binding munc18-1, a middle phosphotyrosine-binding (PTB) domain, and two C-terminal domains. The latter domains have been termed postsynaptic density-95/discs-large tumor suppressor gene/zona occludens-1 (PDZ; PDZa and PDZb) domains (Okamoto and Südhof, 1997). The postsynaptic density-95 protein interacts with the N-methyl-d-aspartate receptor and K+ channels, a step important for their clustering at the plasma membrane (Kim et al., 1995; Kornau et al., 1995). Phosphotyrosine-binding domain is known to mediate assembly of signaling complexes by binding specifically to tyrosine-phosphorylated proteins (Kavanaugh et al., 1995). The PTB domain of mint1 binds phosphatidylinositol phosphates that are required for exocytosis (Okamoto and Südhof, 1997) and an -NPxY-motif in peptides that may not be phosphorylated (Borg et al., 1996). Although the precise function of these domains remains unclear, they mediate interactions with short peptide sequences of target molecules, thereby operating as protein–protein binding domains (Harrison, 1996).
Because no proteins other than mints are known to contain PTB and PDZ domains in one molecule, a unique role of mints in presynaptic terminals appears likely. It has been reported that mint1, a neuronal isoform, is involved not only in synaptic vesicle exocytosis but also in membrane organization in the nerve terminals (Butz et al., 1998; Kaech et al., 1998; Rongo et al., 1998). Recently, the authors reported that mint1 was highly concentrated in the presynaptic active zone (Okamoto et al., 2000). In the current study, the authors examined changes in mint1 that might be related to delayed neuronal death in mouse hippocampus after transient global ischemia.
MATERIALS AND METHODS
Transient global ischemia
Adult male C57B/L6 mice (purchased from Japan SLC, Shizuoka, Japan) 8-to 12-weeks-old and weighing 22 to 28 g were used. During 0.5% to 2% halothane inhalation anesthesia, transient global cerebral ischemia was produced by occluding both common carotid arteries with aneurysm clips for 15 minutes. Using a heat lamp body temperature was maintained at 36.5°C during the operation. Postoperatively, the animals were kept in an incubator at 37°C until active voluntary movement had recovered. To assure similar ischemic insults in each animal, the authors monitored the change in cerebral blood flow in both hemispheres throughout the operation. To accomplish this, an acrylate tube was attached to the intact skull according to stereotactic coordinates (1 mm anterior and 4.5 mm lateral to the bregma), and cerebral blood flow was measured by laser–Doppler flowmetry using a linear probe (diameter, 1 mm; Neuroscience, Tokyo, Japan) inserted into the rigid plastic tube. All operative procedures were completed within 15 minutes.
Immunohistochemistry for mint1
Mice studied immunohistochemically were fixed by perfusion with 4% paraformaldehyde solution in 100 mmol/L phosphate buffer (pH 7.4) at 1, 3, 5, or 7 days after bilateral common carotid artery occlusion (n = 5 for each group). After removal, brains were sectioned in 20-μm thicknesses using a vibratome (Leika VT 1000S; Leica, Heiderberg, Germany). After washing with phosphate-buffered saline (PBS; pH 7.4), all sections were treated with 0.1% H2O2 in PBS to quench endogenous peroxidase and then incubated for 30 minutes in 5% normal bovine serum plus 1.5% normal goat serum. Sera were diluted in PBS containing 0.1% Triton X-100. Immunostaining then was performed overnight at 4°C with rabbit polyclonal antibodies against mint1, munc18-1 (Okamoto and Südhof, 1997), and synaptophysin 2 (Syp2;Castillo et al., 1997). The antibody for mint1 was raised against the N-terminal portion (71a.a. to 439a.a.) of mint1 proteins, which represent the domain interacting with munc18-1 but not the PTB or PDZ domains. After reaction with respective primary antibodies, immunodetection was performed using the Vectastain ABC Elite Kit (Vector, Burlingame, CA, U.S.A.) followed by reaction product visualization using diaminobenzidine. The specificity of the mint1 antibody was confirmed by absorption with an excess of the peptide antigen.
Western blotting for mint1
For Western blotting, mice were decapitated 1, 3, and 7 days after common carotid artery occlusion and at 3 days after sham ischemia (n = 3 for each group). Both hippocampi from 3 animals were homogenized and lysed in a 10-fold excess (w/v) of PBS containing 1% Nonidet-P, 1% phenylmethylsulfonyl fluoride, and 5 mmol/L EDTA. For comparison, small portions of cerebral cortex from the same animals were processed similarly. After centrifugation (10,000×g for 15 minutes at 4°C), the protein concentration of the supernatant was determined using a protein assay kit (Bio-Rad, Hercules, CA, U.S.A.). The mixture (equivalent to 30 μg protein) was boiled for 5 minutes with 20 μL of sodium dodecyl sulfate gel-loading buffer (62.5 mmol/L Tris-HCl, 2% sodium dodecyl sulfate, 0.01% bromophenol blue, 10% glycerol, and 5% 2-mercaptoethanol) and was subjected to polyacrylamide gel electrophoresis (polyacrylamide concentration, 7.5%). After transfer by blotting of proteins from gels to nitrocellulose membranes, the membranes were treated with skim milk for 1 hour at room temperature to block nonspecific binding of antibodies. Membranes then were probed overnight at 4°C with the primary antibodies at a dilution of 1:300 in 10% (v/v) skim milk and 0.05% (v/v) Tween 20 in PBS. Filters then were washed 3 times for 15 minutes in 0.1% Tween 20 in PBS, then incubated for 2 hours at room temperature with horseradish peroxidase-conjugated goat anti-rabbit IgG (Cappel, West Chester, PA, U.S.A.) at a dilution of 1:1500 in PBS containing 0.05% (v/v) Tween 20. Immunoreactive bands were visualized by enhanced chemoluminescence (Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.).
RESULTS
Localization of mint1 in hippocampus
In the hippocampus of mice with sham ischemia (n = 3), a heterogeneous, region-specific distribution of finely granular reactive product was observed, representing mint1 immunoreactivity (Fig. 1A). Pyramidal neurons of the CA sectors, granule cells of the dentate gyrus, and their dendrites showed a negative pattern. However, some scattered, intensely stained neurons were observed throughout the hippocampus, especially in the dentate subgranular proliferative zone. Granular deposits representing immunoreactivity were abundant in the entorhinal projection area of the hippocampus (Gaarskjaer, 1986). More specifically, the mint1 immunoreactivity was strong in the lacunosum moleculare in Ammon's horn, where the apical dendrites receive input from the entorhinal area. In the molecular layer of the dentate gyrus, staining was strong in the two distal portions, but the proximal portion close to the granule cells, which is termed the commisural system, was only weakly stained (Fig. 1A). In addition, mint1 immunoreactivity was strong in the mossy fiber projection, the neural connection between the granule cells of the dentate gyrus and CA3 pyramidal neurons (Fig. 3A). No small cells with the appearance of glia were stained anywhere in the brain.
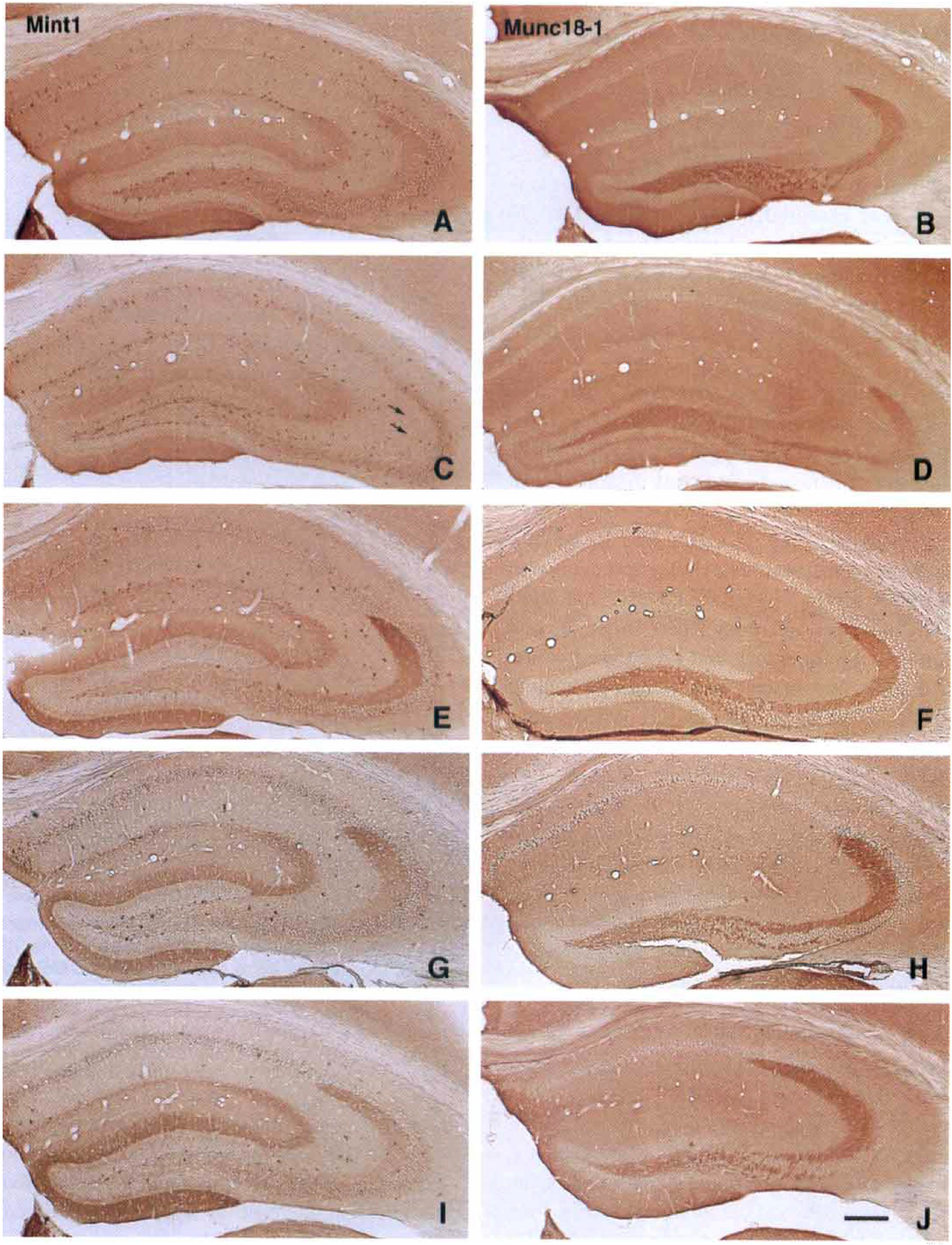
Distribution and time course of immunoreactivity for mint1 (Mint1;
Time course of mint1 expression after ischemia
The authors compared mint1 expression in postischemic mice with that of two other synaptic proteins, munc18-1 and Syp2, using adjacent hippocampal sections. In mice with sham ischemia, munc18-1 was strongly expressed in the mossy fibers; in other regions its distribution was essentially uniform (Fig. 1B). In sham-ischemic mice, Syp2 was identified predominantly in the mossy fiber bundle and was present at lesser levels in the proximal region of the molecular layer of the dentate gyrus, as described previously (Castillo et al., 1997; Fig. 2A).
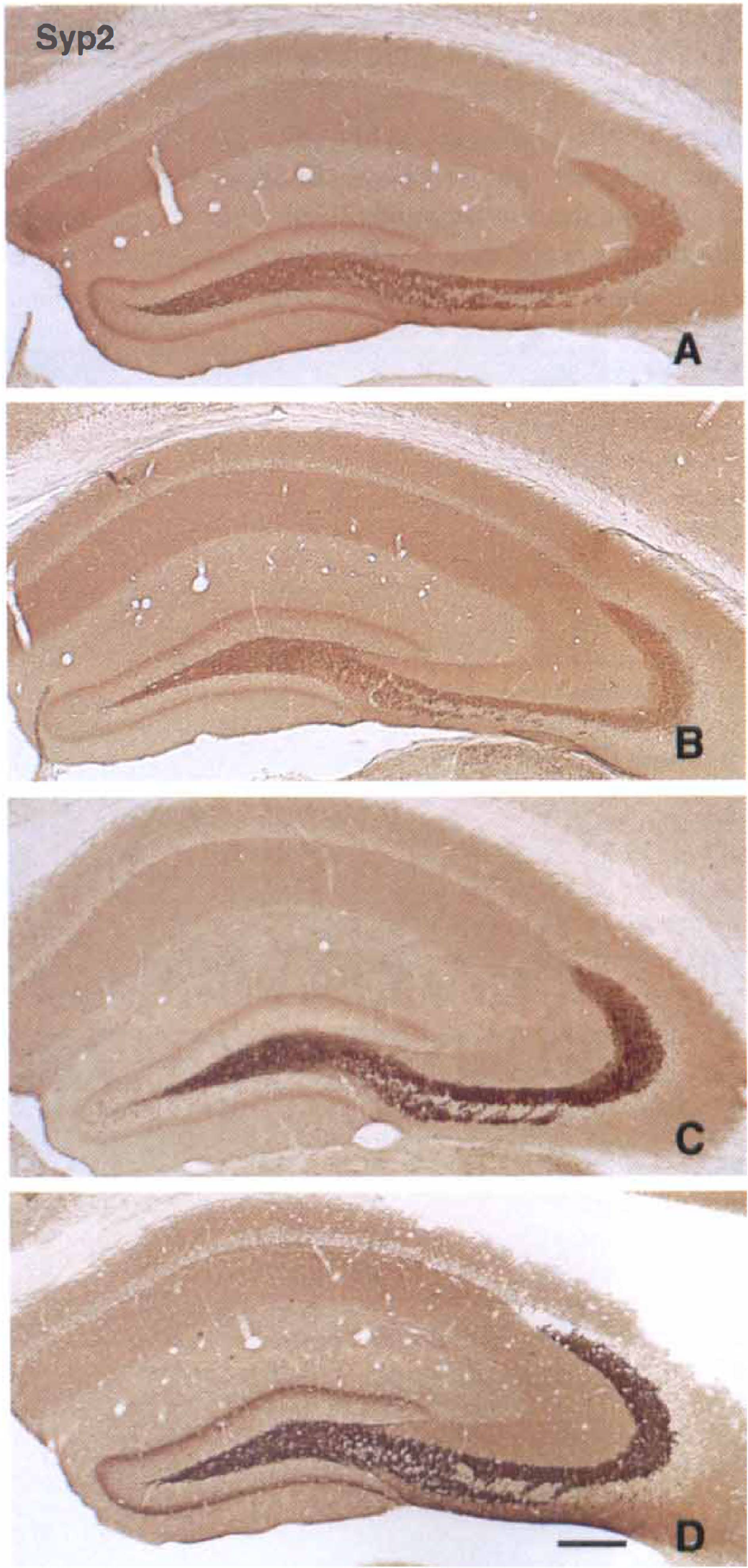
Distribution and time course of immunoreactivity for synaptophysin 2 (Syp2) in mouse hippocampus after 15 minutes of ischemia. Sections were prepared from control mouse
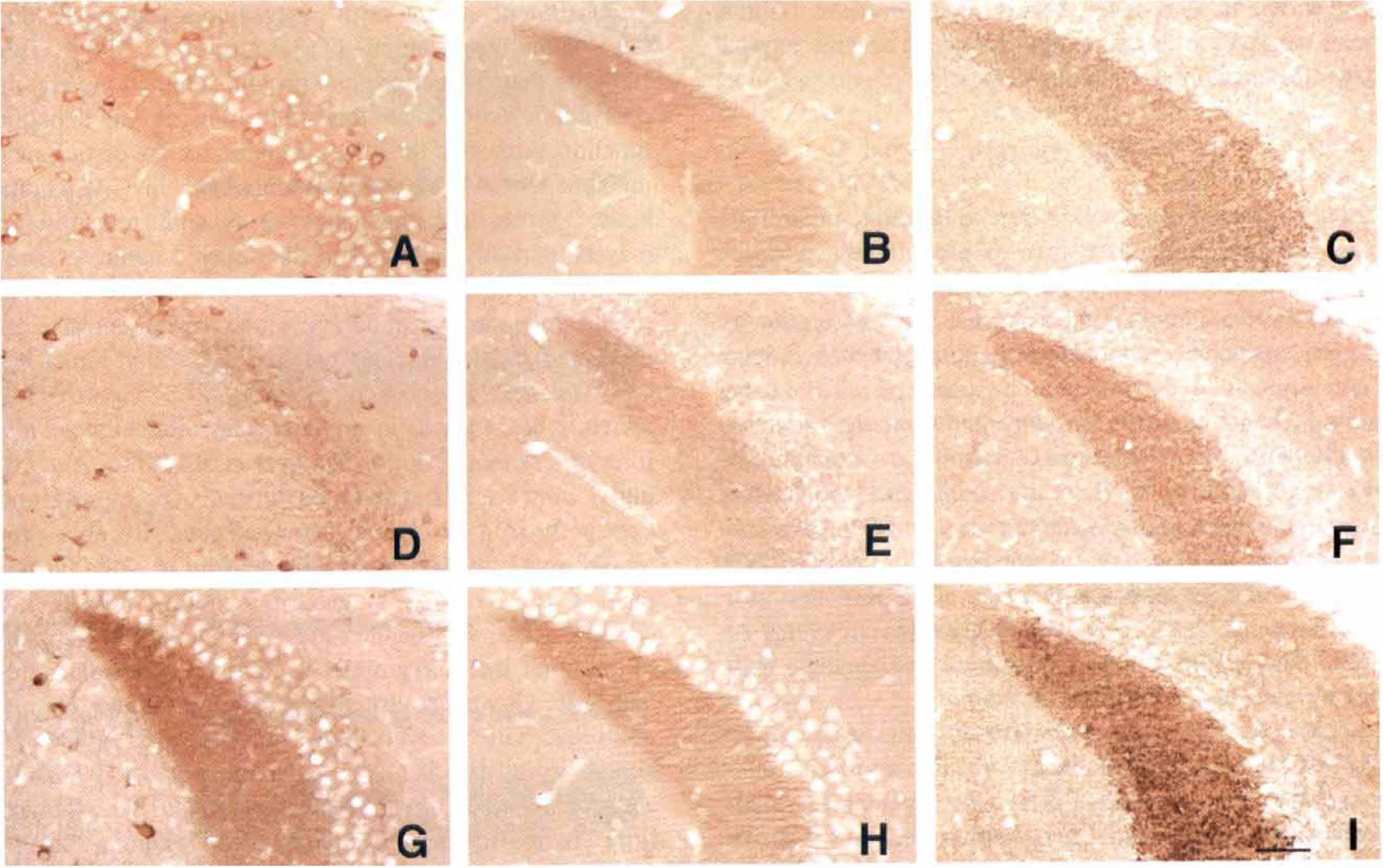
Higher magnification of Figs. 1 and 2 representing the time course of immunoreactivity for mint1
After 1 day of reperfusion, ischemic mice generally showed mildly diminished immunostaining for all synaptic proteins in mossy fibers (Fig. 3D to 3F). Mint1 expression in mossy fibers had selectively disappeared at 1 day of reperfusion, whereas the other 2 proteins were substantially expressed (Figs. 1C, 1D, 2B, 3D to 3F). After 3 days of reperfusion, munc18-1 expression was restored to control levels (Figs. 1F and 3H) and Syp2 expression was increased in mossy fibers (Figs. 2C and 3I). Immunoreactivity for mint1 had reappeared in the mossy fiber bundle, and in fact was enhanced beyond control levels at 3 days (Figs. 1E and 3G). After 5 days of reperfusion, delayed neuronal death was observed in CA1 pyramidal neurons, and expression of mint1 was decreased in the stratum radiatum of the CA1 sector (Figs. 1G and 4C), whereas munc18-1 was expressed in the same region (Figs. 1H and 4D). After 7 days of reperfusion (Figs. 1I, 1J, 2D), these 3 proteins were expressed in the mossy fiber bundle. In the stratum radiatum, however, mint1 expression was no longer detected, whereas the other two proteins were expressed. All samples of ischemic brain stained with mint1 antibody in the presence of an excess of the peptide used as the immunogen showed no staining (not illustrated), indicating that staining of mint1 was specific.
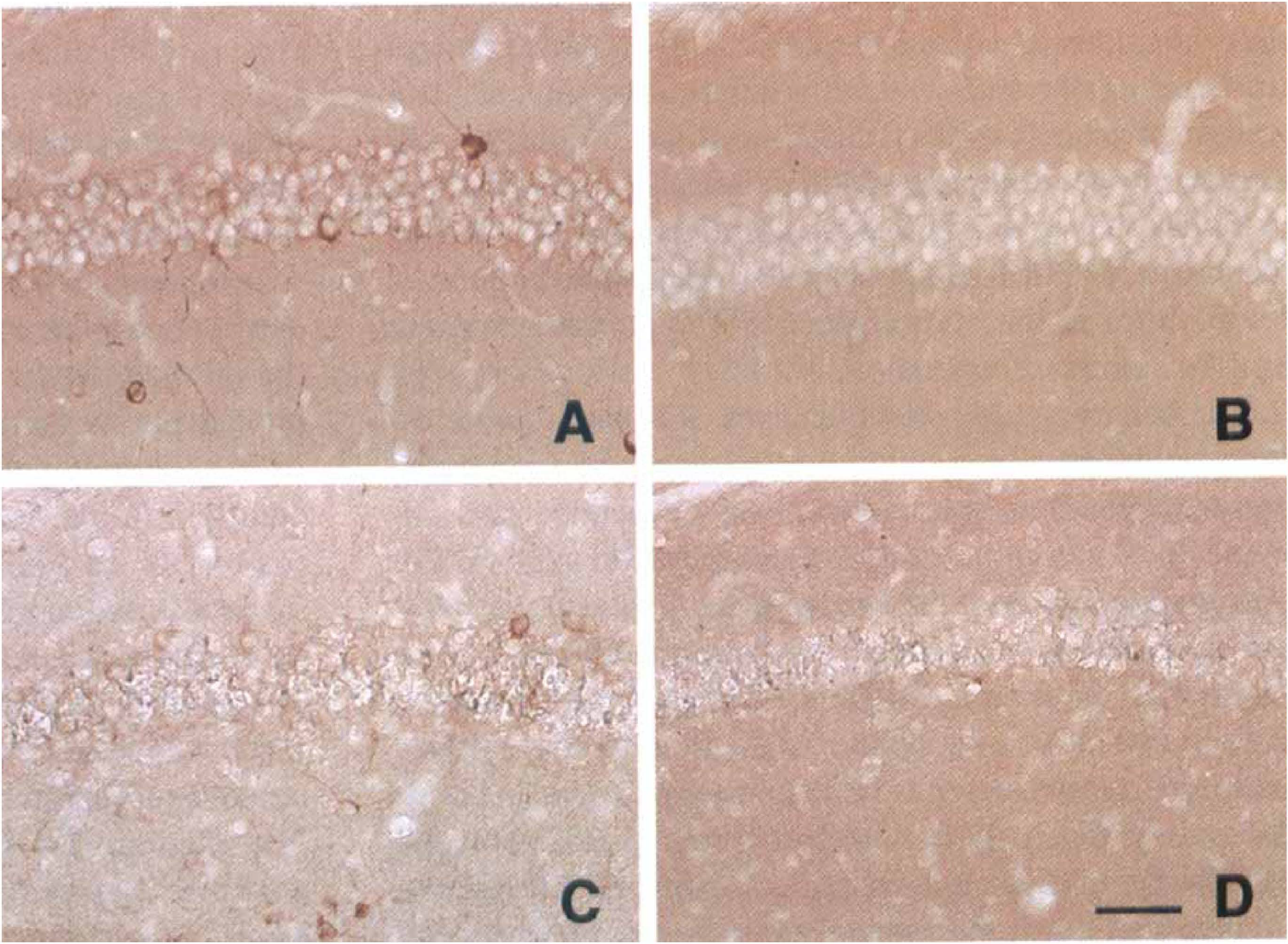
Higher magnification of Fig. 1, representing the change in immunoreactivity for mint1
To determine whether de novo synthesis of mint1, munc18-1, and Syp2 proteins changed after ischemia, Western blotting was performed on homogenates from the hippocampus (Fig. 5A) and cerebral cortex (Fig. 5B). An immunoreactive species corresponding to 140 kDa (the size of mint1), 68 kDa (the size of munc18-1), or 37 kDa (the size of Syp2) were detected in normal brains. Expression of these three proteins in cerebral cortex was relatively stable during the entire reperfusion period (Fig. 5B). In the hippocampus, mint1 expression was increased slightly at 3 days and was decreased at 7 days, and a slight increase in Syp2 was observed at 3 days of reperfusion (Fig. 5A). In contrast, expression of munc18-1 protein did not change appreciably during the entire reperfusion period. The reduction of mint1 in mossy fibers shown by immunohistochemistry could not be detected by this method, most likely because the immunoreactivity of other components such as neurons was not influenced after ischemia (Fig. 3D).
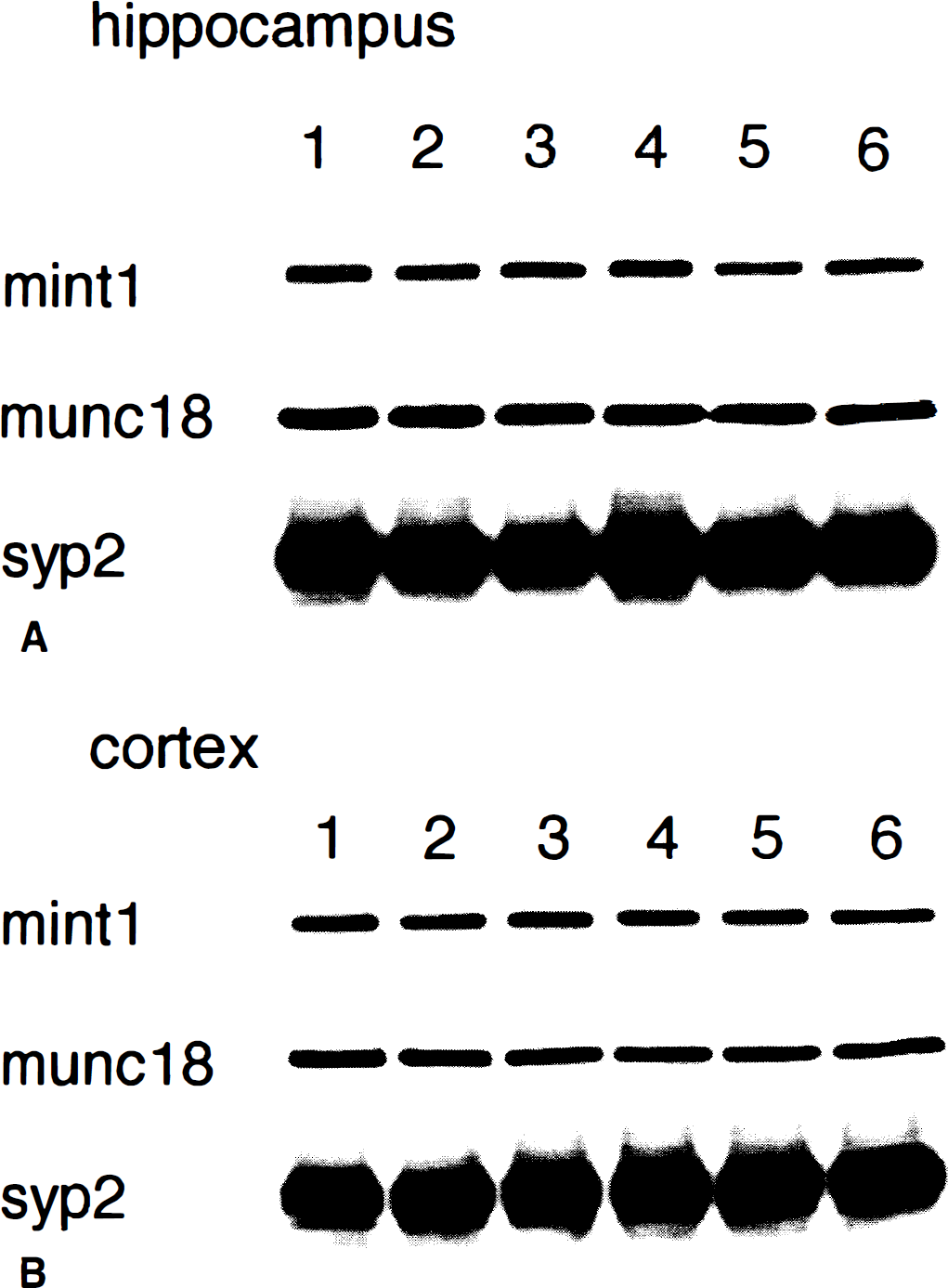
Immunoblotting of mint1, munc18-1, and synaptophysin 2 (syp2) in the mouse hippocampus
DISCUSSION
The current study demonstrated that unlike other synaptic proteins, mint1 is expressed predominantly in the entorhinal projection. This distribution suggests that mint1 plays an important role in neuronal transmission in the excitatory projection from the entorhinal cortex to the dentate gyrus. In addition, the presence of mint1 in the mossy fiber projection, the only known neural connection between the dentate area and Ammon's horn (Gaarskjaer, 1986), suggests that mint1 can affect information transfer between the entorhinal cortex and CA3 pyramidal neurons. Although the neurotransmitter specificity of synaptic vesicle interactions involving mint1 is not known, mint1 and other synaptic proteins are likely to participate in central neurotransmission either regionally or in a neuron-specific manner.
The synaptic vesicle cycle includes nine steps: docking, priming, fusion and exocytosis, endocytosis, translocation to the interior, endosome fusion, budding, neurotransmitter uptake, and translocation back to the active zone (Südhof, 1995). Among these steps, exocytosis by membrane fusion is the key event in vesicle cycle. Mint1, interacting with the munc18-1/syntaxin complex, may contribute to synaptic vesicle exocytosis (Okamoto and Südhof, 1997). Another set of ternary complex, CASK/velis/mint1, has been proposed to participate in polarized localization of membrane proteins in neurons (Butz et al., 1998; Kaech et al., 1998). Somatodendritic localization of mint proteins with CASK has been reported (Borg et al., 1999). Thus, mint1 might have multiple functions in neurons depending on its partners, and the function of mint1 where it coexists with munc18-1 in presynaptic boutons may differ from its role in the cell soma or in dendrites. In addition, because codistribution of mint1 with munc18-1 was essentially limited to certain areas within the hippocampus, neurotransmitter release through the mint1/munc18-1/syntaxin complex may be regionally specific.
The current study sought changes in synaptic proteins that could affect neuronal transmission after an ischemic episode. Because immunoreactivity of mint1, but not munc18-1 or Syp2, showed transient decreases in the presynaptic region of the mossy fiber bundle, these synaptic proteins may be differentially regulated after ischemia. Consequently, an impairment of the synaptic vesicle cycle because of the lack of mint1 may occur with incomplete exocytosis in presynaptic terminals of the mossy fibers. It has been reported that deficiency of complexin II, a presynaptic protein that is directly involved in synaptic vesicle exocytosis, impaired the establishment of long-term potentiation in the CA1 and CA3 of the hippocampus (Takahashi et al., 1999). This suggests that correct protein–protein interactions among presynaptic proteins are important in regulating synaptic function. Although the direct influence of the disturbance in input to CA3 on the neuronal death in CA1 is unknown, a transient impairment of synaptic transmission may be partially involved in the delayed nature of neuronal death in CA1.
The mechanism underlying the disappearance of mint1 remains to be investigated. Structurally, mint1 has a possible PEST sequence (a.a. 3-31, PEST Score 5.86), which is often found in proteins with intracellular half-lives of less than 2 hours (Rodgers et al., 1986). Accordingly, mint1 is thought to be turned over rapidly and constantly in nonischemic neuronal cells. Because protein synthesis in CA1 pyramidal neurons is globally suppressed in the early stages of ischemia–reperfusion (Kirino, 1982; Widmann et al., 1991), a reduced amount of proteins including mint1 is likely in neurons after ischemia. However, protein synthesis as shown by immunoreactivity recovers more quickly in CA3 and the dentate gyrus than in CA1 (Matsuyama et al., 1993a). In addition, another synaptic protein, munc18-1, showed little change in immunostaining after ischemia. Therefore, reduced mint1 immunoreactivity may not simply reflect suppressed protein synthesis, or impaired axoplasmic transport from neuronal perikarya, or both; the change may imply greater vulnerability of mint1 molecules to ischemia. Mint1 has several possible cleavage sites of calpains, one of major calcium-dependent proteases. Because calpains are reported to be activated by ischemic stress (Lipton, 1999), it is suggested that mint1 could be degraded easily by calpains after ischemia. The authors also have found the degradation of mint1 in a calcium-dependent manner in vitro (Okamoto, unpublished data). Instead, the vulnerability may reflect the difference in the intracellular localization of this molecule. Recently, the authors have shown the ultrastructural localization of mint1, munc18-1, and Syp2, in which mint1 was highly concentrated in the presynaptic active zone, whereas munc18-1 was enriched throughout the presynaptic membrane, and Syp2 was only detected on synaptic vesicles (Okamoto et al., 2000).
The current study showed the enhancement of mint1 and Syp2 in the mossy fiber bundle at 3 days of reperfusion. It has been reported that a synaptosomal-associated protein, SNAP-25, involved in neurotransmitter release, is increased in the mossy fibers as early as at 2 days of reperfusion in the gerbil model (Martí et al., 1998). Thus, it is suggested that an increase in some but not all presynaptic marker proteins may occur in mossy fibers before the delayed neuronal death has been detected in CA1. A late increase in mint1 immunoreactivity in mossy fibers probably involves protein synthesis. Activation of protein synthesis in dentate granule cells has been reported in rodent models of ischemia (Matsuyama et al., 1993a, 1993b, 1994; Tagaya et al., 1995). Regional induction of mint1 mRNA in dentate granule cells should be investigated by in situ hybridization to confirm this finding.
A number of studies have demonstrated a contribution of increased synaptic transmission in Schaffer collaterals to delayed neuronal death in the CA1 sector (Storm-Mathisen, 1981; Onodera et al., 1986; Westerberg et al., 1989; Benveniste et al., 1989; Meldrum, 1990; Choi and Rothman, 1990). However, glutamate efflux through Schaffer collaterals does not appear directly linked to a regional susceptibility that characterizes ischemic damage in CA1. First of all, glutamate released into the extracellular space during ischemia is rapidly cleared during reperfusion (Obrenovitch and Richards, 1995). Second, extracellular glutamate concentrations are increased in CA1 and the more resistant CA3 field of the hippocampus during ischemia (Mitani et al., 1992). In addition, electrophysiologic studies have found that CA3 neurons, which provide the major excitatory input to CA1, are relatively silent during reperfusion, whereas CA1 neurons are dying (Chang et al., 1989; Urban et al., 1989; Kirino et al., 1992). These results suggest that delayed neuronal death results from an intrinsic disturbance in CA1 neurons rather than altered excitation in the hippocampal circuitry. Nonetheless, an intact pathway from the dentate gyrus that provides glutamatergic input to CA1 pyramidal neurons probably is required for delayed neuronal death to occur, even though synaptic transmission may not be increased. Prior lesions affecting hippocampal circuitry partially protect CA1 pyramidal neurons from ischemic degeneration (Johansen et al., 1987; Kaplan et al., 1989).
Whether postischemic events related to glutamatergic synaptic systems include both postsynaptic and presynaptic changes has not been fully investigated. In CA1, evidence has accumulated indicating that presynaptic components are more resistant to ischemia than postsynaptic structures (Johansen et al., 1984; Kirino et al., 1990; Kitagawa et al., 1989, 1992; Yanagihara et al., 1990; Martíet al., 1998). However, Von Lubitz and Diemer (1983) demonstrated in an electron microscopic study that synaptic vesicles are considerably reduced in the active zones of boutons in the rat CA1 sector after 10 minutes of global ischemia. Furthermore, Miyazawa et al. (1995) described a transient decrease in immunoreactivity of a synaptic vesicle-associated protein during an early stage of the neuronal death in CA1. Thus, an ischemic insult is likely to affect, even if transiently, synaptic vesicles and postsynaptic structures in the CA1 region. The loss of target cells may trigger reactive changes in Schaffer collaterals, which may result in variations in the pattern of immunostaining of presynaptic markers (Miyazawa et al., 1995; Martí et al., 1998). The current study demonstrated that, unlike other synaptic proteins, mint1 was not expressed in the stratum radiatum of the CA1 sector exhibiting the neuronal death. This finding also suggests that mint1 is no longer detected in the presynaptic active zone when the postsynaptic structures have been disrupted. The authors' study of postischemic expression of mint1, munc18-1, and Syp2 in the hippocampus may offer a new perspective regarding the pathogenesis of delayed neuronal death.
In conclusion, the late induction of mint1 during reperfusion may be concomitant with late restoration of the excitatory pathway to the hippocampus. This chronologic pattern may involve possible protein–protein interactions among the presynaptic proteins affected by ischemia, and this type of synaptic disturbance is likely to be involved in the process of delayed neuronal death. However, exocytotic and nonexocytotic modes of neurotransmitter release has been reported during chemical ischemia in vitro (Pocock and Nicholls, 1998). Further investigation on the relation between neurotransmitter release and synaptic proteins after cerebral ischemia is needed.