
Abstract
Mesenchymal stromal cells (MSCs) are viewed as immune-privileged cells and have been broadly applied in allogeneic adoptive cell transfer for regenerative medicine or immune-suppressing purpose. However, the surface expression of human leukocyte antigen (HLA) class I molecules on MSCs could still possibly induce the rejection of allogeneic MSCs from the recipients. Here, we disrupted the β2 microglobulin (B2M) gene in human peripheral blood mononuclear cell-derived induced pluripotent stem cells (iPSCs) with two clustered regulatory interspaced short palindromic repeat (CRISPR)-associated Cas9 endonuclease-based methods. The B2M knockout iPSCs did not express HLA class I molecules but maintained their pluripotency and genome stability. Subsequently, MSCs were derived from the HLA-negative iPSCs (iMSCs). We demonstrated that B2M knockout did not affect iMSC phenotype, multipotency, and immune suppressive characteristics and, most importantly, reduced iMSC immunogenicity to allogeneic peripheral blood mononuclear cells. Thus, B2M knockout iPSCs could serve as unlimited off-the-shelf cell resources in adoptive cell transfer, while the derived iMSCs hold great potential as universal grafts in allogeneic MSC transplantation.
Introduction
Stem cell-based therapy in a form of cell transplantation plays a key role in living-cell medicine nowadays. Such cell transplantation requires an adoptive transfer of certain types of therapeutic stem cells or cell derivatives. Cell derivatives from pluripotent stem cells and some lineage-specific multipotent stem cells are administrated. Among the multipotent stem cells for transplantation, mesenchymal stem cells (MSCs), which are also called mesenchymal stromal cells, are extensively studied and applied in versatile therapies. These MSCs are given birth in embryonic germ layers and located in adult tissues as cell reservoirs to support local tissue repairing with the ability of differentiation into adipocytes, osteocytes, chondrocytes, 1 and even myocytes. Besides, MSCs are found immunosuppressive as they could suppress T cell proliferation by secreting inhibitory factors, like indolamine 2,3-dioxygenase (IDO), transforming growth factor-beta (TGF-β), and hepatocyte growth factor (HGF), and directly forming inhibitory ligation through cell-to-cell contact 2,3 . With the above characteristics, the adoptive transfer of MSCs has been conducted in treating many diseases, including refractory wounds 4 , osteogenesis imperfecta, 5 and myocardial infarction 6 . Multiple clinical trials were also performed to delve into the therapeutic effects of MSCs on osteoarthritis of the knees 7,8 , acute and chronic graft versus host diseases (GvHDs) 9 –11 , and immune disorders 12 . Some of the clinical trials had already involved allogeneic MSCs due to the privilege of broad immune suppression from MSCs to both host and graft immune cells.
In practice, MSCs can be isolated from human adipose tissue, bone marrow, and umbilical cord. With regard to clinical application, most of the MSCs were isolated and expanded from patients themselves or healthy donors, but those cell resources are limited and heterogenous among donors. In this case, pluripotent stem cells are considered as potent and homogenous cell resources to generate MSCs. Many studies have investigated the differentiation from pluripotent stem cells to MSCs, while human-induced pluripotent stem cells (hiPSCs) are the ideal source free of an ethical issue. MSCs generated from human iPSCs have shown similar differentiation capacity and immunosuppressive properties as the primary MSCs 13 –15 . Furthermore, a clinical trial of iPSC-derived MSCs treating acute GvHDs has been ongoing, which was approved by UK Regulatory Authority.
Although hiPSC-derived MSCs were reported with immune privileges as an allogeneic graft, immune responses against those allogeneic MSCs were still a concern in adoptive MSCs transfer 16,17 . According to previous studies, MSCs could upregulate their human leukocyte antigen (HLA) expression after the exposure to cytokine stimulation such as interferon-gamma (IFN-γ) 18 . Such upregulation of HLA molecules could provoke the adaptive immune responses, including antibodies and cytotoxic memory T cells against graft MSCs. Adaptive memory T cells were observed in animal models of allogeneic MSCs infusion, indicating the potential cellular rejection of those allogeneic MSCs 19 . Based on the above findings, although no adverse record was spotted from over thousands of patients in allogeneic MSCs clinical trials so far 17 , rejection of the allogeneic MSCs could still occur. Rejection of MSCs was only much slower than the rejection of other allogeneic graft tissues 16 . To further reduce the immunogenicity of allogeneic hiPSC-derived MSCs, B2M knockout (B2MKO) was proposed as a solution to achieve the hypoimmunogenic hiPSCs for MSC generation.
Here, we explored the B2MKO in human peripheral blood mononuclear cell (PBMC)-derived iPSCs by clustered regulatory interspaced short palindromic repeat (CRISPR)-associated Cas9 endonuclease (CRISPR/Cas9) system and generated several B2MKO hP-iPSCs single-cell lines. These B2MKO hP-iPSC lines maintained their pluripotency and genome stability. Afterward, we generated functional MSCs from those B2MKO hP-iPSCs. These B2MKO hP-iPSC-derived MSCs demonstrated equivalent multipotency but lower immunogenicity to allogeneic PBMCs.
Materials and Methods
Cell Cultures
Human PBMC-derived iPSCs were reprogrammed from healthy donor’s PBMCs using Epi5 Episomal iPSC Reprogramming Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. hP-iPSCs were selected as single-cell clones and characterized as described previously
20
. hP-iPSCs were cultured in mTeSR1 medium (STEMCELL Technologies, Vancouver, Canada) on Matrigel hESC-qualified Matrix (BD Biosciences, Franklin Lakes, NJ, USA) coated plates. Culture medium was refreshed every day, while hP-iPSCs were subcultured every week. To form embryoid bodies from hP-iPSCs, confluent iPSC colonies were dissociated with 1 mg/ml Dispase (Thermo Fisher Scientific) as large cell aggregates. Those cell aggregates were cultured in low attachment plate with embryoid body (EB) medium as Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F12, Gibco, Thermo Fisher Scientific) supplemented with 20% KnockOut serum replacement (Gibco), 2 mM
MSCs were differentiated from hP-iPSCs directly as described previously
21
. When hP-iPSCs reached confluence, the culture medium was replaced by DMEM with low glucose (Gibco), 10% fetal bovine serum (FBS, Hyclone GE Healthcare, Little Chalfont, UK), and 2 mM
Fresh PBMCs were isolated from a healthy donor’s buffy coat with Ficoll-Paque PREMIUM 1.084 (GE Healthcare) by density gradient centrifugation. PBMCs were primed with MSCs by co-culture in AIM V Medium (Gibco) with 5% human AB serum (Valley Biomedical, Winchester, VA, USA) and 300 IU/ml interleukin-2 (IL-2; PeproTech, Rocky Hill, NJ, USA) for T cell activation. Activated CD3+ T cells were restimulated with MSCs at a ratio of 5:1 every 7 days to achieve the population of MSC-specific cytotoxic T lymphocytes (CTLs). Primary natural killer (NK) cells were expanded from fresh PBMCs with inactivated modified K562 cells as feeder cells 22 . The expanded NK cells were collected 7 to 10 days after co-culture with K562 cells as a purity of more than 90% CD3−CD56+ cells for cell cytotoxicity assay.
Plasmid Construction
The pX260 and pX459 plasmids (Addgene, Cambridge, MA, USA) containing a CRISPR/Cas9 system, which was described previously 23,24 , were used in this study. Target sequences of CRISPR/Cas9 were designed by CHOPCHOP (http://chopchop.cbu.uib.no/) and listed in Table S1. The B2M Target1 was subcloned into pX260, while the B2M Target2 was subcloned into pX459. Two donor plasmids for B2M Exon1 homology-direct integration were constructed. One was designed with a eukaryotic translation elongation factor 1α (EF1α) promoter driving the expression of an EGFP gene and a mouse phosphoglycerate kinase 1 (PGK) promoter driving the expression of the Neo gene (the neomycin resistant gene), while the other was designed with the EF1α promoter driving the expression of a mCherry gene and a Simian virus 40 (SV40) promoter driving the expression of the Hygro gene (the hygromycin resistant gene). Both donor sequences were flanked by homologous DNA sequences from B2M EX1 locus (chromosome 15: nucleotides 44,710,501–44,711,401 and nucleotides 44,711,615–44,712,485, GRCh38.p2 Primary Assembly).
Generation of B2 M Knockout hP-iPSC Clones
B2MKO hP-iPSCs were generated by two methods: double-color selection and one-shot puromycin selection. For the double-color selection, hP-iPSCs were dissociated by Accutase (Merck Millipore), washed by phosphate-buffered saline (PBS, Lonza), and resuspended in Opti-MEM I Reduced Serum Medium (Gibco) as single-cell suspension; 1 × 106 cells were transfected with 2.5 µg pX260 plasmid and 2.5 µg donor plasmid with GFP by electroporator (Nepa Gene, Chiba, Japan). Transfected single cells were recovered in NutriStem hPSC XF Medium (Biological Industries, Beit-Haemek, Israel) and seeded on Matrigel-coated plates. Four days after electroporation, the culture medium was changed back to mTeSR1 medium, and the cells were selected by 25 μg/ml Geneticin (G418 Sulfate, Gibco) for 2 weeks. Selected cells were subjected to fluorescence-activated cell sorting (FACS) for single-cell seeding, which was performed by BD FACSAria I Flow Cytometer (BD Biosciences). Selected hP-iPSCs were dissociated by Accutase as single cells, and the GFP positive population was seeded as one cell per well on Matrigel-coated 96 well-plates in the NutriStem medium. Single-cell clones were expanded, genotyped by PCR and sequencing. A monoallelic knockout single-cell clone was confirmed and preceded to a second round of knockout on the other allele. The monoallelic knockout single-cell clone was transfected with 2.5 µg pX260 plasmid and 2.5 µg donor plasmid with mCherry by electroporation. Transfected cells were selected by 10 μg/ml Hygromycin B (Gibco) for 2 weeks and subjected to single-cell seeding as well. The double-color single-cell clones were collected, expanded, and confirmed by genotyping.
For the one-shot puromycin selection, 1 × 106 hP-iPSCs were transfected with 5 µg pX459 plasmid by electroporation and recovered in the NutriStem medium on Matrigel-coated plates overnight. Afterward, transfected cells were selected by 1 μg/ml puromycin (Thermo Fisher Scientific) in the NutriStem medium for 24 h. Survival single cells were cultured in fresh Nutristem medium for 3 to 4 days before changed back to mTeSR1 culture. Single-cell clones were isolated and subjected to genotyping and phenotyping. B2M negative clones were confirmed from those single-cell clones.
Western Blotting and Flow Cytometry Analysis
For Western blot analysis, sample proteins were extracted by lysing cells with radioimmunoprecipitation assay (RIPA) buffer (Nacalai Tesque, Kyoto, Japan), analyzed in sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel under reducing condition, and then electroblotted to a nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA, USA). Rabbit Anti-B2M antibody clone EP2978Y (1:5000 dilution, Abcam, Cambridge, UK) and mouse anti-β-actin antibody clone GT5512 (1:1000 dilution, Abcam) were used as primary antibodies. Goat anti-rabbit immunoglobulin G (IgG)-horseradish peroxidase (HRP) (1:5000 dilution, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and goat anti-mouse IgG-HRP (1:2000 dilution, Santa Cruz Biotechnology) were used as secondary antibodies. The membrane was developed and visualized for chemiluminescence by MYECLImager (Thermo Fisher Scientific). For flow cytometry analysis, iPSCs and MSCs were stained with antibodies in autoMACS Running Buffer (Miltenyi Biotec, Bergisch GladBach, Germany) and analyzed by BD Accuri C6 Flow Cytometer (BD Biosciences). Data were analyzed by CFlow Sampler software (BD Biosciences) and antibodies in flow cytometry assays were listed in Table S2.
Southern Blotting
Southern blot was performed as described previously 25 . For each sample, 15 µg genomic DNA was digested with 50 U HindIII-HF (New England Biolabs, Ipswich, MA, USA) overnight. Digested DNA was loaded on a 1% agarose gel, and gel electrophoresis was performed at 40 V for 5 h. DNA was then transferred to a positively charged nylon membrane by using iBlot Dry Blotting System (Invitrogen, Thermo Fisher Scientific). The membrane was washed with 1.5 M sodium chloride/0.5 M sodium hydroxide denaturing solution and then air-dried. Ultraviolet cross-linking was performed at 130 mJ/cm2. The membrane was first prehybridized in DIG Easy Hyb (Roche Diagnostics, Basel, Switzerland) buffer for 1 hour and then hybridized with a DIG-labeled probe overnight. Afterward, the membrane was first washed twice with 2× saline-sodium citrate (SSC)/0.1% sodium dodecyl sulfate (SDS) at 40 °C and then twice with 0.1× SSC/0.1% SDS at 50 °C. Then, the membrane was blocked and washed with DIG Wash and Block Buffer Set (Roche Diagnostics) and incubated with an anti-digoxigenin-AP (Roche Diagnostics). Finally, the membrane was detected with CDP-Star (Roche Diagnostics) as a substrate for chemiluminescence by using MYECL Imager (Thermo Fisher Scientific). The probes were synthesized by using the PCR DIG Probe Synthesis Kit (Roche Diagnostics) with donor plasmid for the B2M gene as a template. Primers for probe synthesis were listed in Table S1.
In Vitro Differentiation of hP-iPSC-Derived MSCs
For adipogenesis, iPSC-derived MSCs were seeded at a density of 10,000 cells/cm2 for 2 to 4 days to reach confluence. Then culture medium was changed to differentiation medium from StemPro Adipogenesis Differentiation Kit (Gibco) and refreshed every 3 to 4 days for more than 2 weeks. Cells were then fixed with 4% formaldehyde (Sigma-Aldrich) and histologically stained by Oil Red O (Sigma-Aldrich) for lipid content.
For osteogenesis, iPSC-derived MSC cells were seeded at a density of 5000 cells/cm2 for 2 to 4 days. Then culture medium was changed to differentiation medium from StemPro Osteogenesis Differentiation Kit (Gibco) and refreshed every 3 to 4 days for more than 3 weeks. Cells were then fixed with 4% formaldehyde (Sigma-Aldrich) and histologically stained by Alizarin Red S solution (Merck Millipore) for calcification.
For chondrogenesis, iPSC-derived MSC cells were concentrated to 1.6 × 107 cells/ml. Cells were loaded onto a plate by 5 µl droplets and cultured as pellets for 2 h to form cell clusters. Then culture medium was changed to differentiation medium from StemPro Chondrogenesis Differentiation Kit (Gibco) and refreshed every 3 to 4 days for more than 3 weeks. Cells were then fixed with 4% formaldehyde (Sigma-Aldrich) and histologically stained by Alcian blue 8GX (Sigma-Aldrich) for acidic polysaccharides.
Reverse Transcription Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated from each cell sample by TRIzol Reagent (Thermo Fisher Scientific). The reverse transcription was performed by SuperScript III First-Strand Synthesis System (Invitrogen). PCR amplification was performed by KAPA Taq ReadyMix PCR Kit (Kapa Biosystems, Roche Diagnostics, Basel, Switzerland) with according annealing temperatures for 35 cycles. The PCR products were resolved by 1.5% agarose gel for analysis. Primers and their relevant annealing temperatures were listed in Table S1.
Mixed Lymphocyte and MSC Reactions (MLMRs)
The MLMRs were performed as described previously 26 . Total 1 × 107 allogeneic PBMCs were labeled by CellTrace Far Red Cell Proliferation Kit (Thermo Fisher Scientific) in Opti-MEM I Reduced Serum Medium. Next, labeled PBMCs were washed with PBS, resuspended in AIM V medium supplemented with 5% human AB serum and 300 IU/ml IL-2, and activated by 50 ng/ml anti-CD3 (OKT-3) antibody. At the same time, these labeled PBMC cultures were mixed with or without hP-iPSC-derived MSCs in a 2:1 ratio. Suspension cells in the mixed cell culture were collected 72 h after reaction for flow cytometry analysis. The dilution of Far Red labeling and the phenotype of activated PBMCs were assessed by flow cytometry assay. For the MLMR assay, more than three independent donors of PBMCs were examined in this experiment.
Cell Cytotoxicity Assay
Cell cytotoxicity was assessed in a standard 2-h Europium-release assay with DELFIA EuTDA Cytotoxicity Reagents (PerkinElmer, Waltham, MA, USA). Assay was performed according to the manufacturer’s instructions. Target cells were first labeled with BATDA reagent at 37 °C for 5 to 15 min and then washed three times with PBS. Effector cells and labeled target cells were mixed in triplicate at different effector to target (E:T) ratios in AIM V medium with 5% human AB serum. Mixed cells were incubated at 37 °C in a humid incubator for 2 h. Spontaneous and maximum releases were determined by incubating target cells without effector cells and with lysis buffer, respectively. After incubation, supernatants were transferred to mix with the Europium solution and analyzed by VICTOR Time-resolved fluorometer (PerkinElmer). The percentage of specific lysis was calculated as:

Immunostaining
EBs were derived from iPSCs and subcultured in the EB medium for 2 weeks. Mature EBs were collected for immunostaining of three germ layers markers, and the staining was performed according to the manufacturer’s instruction of 3-Germ Layer Immunocytochemistry Kit (Thermo Fisher Scientific). In detail, EBs were first fixed and permeabilized at room temperature. Before the staining, EBs were also incubated with a blocking solution for 30 min. After the blocking, primary antibodies were added to stain EBs at 4 °C overnight. Subjected to washing, EBs were then stained with secondary antibodies at room temperature for 1 h. After the second round of washing, stained EBs were visualized by a microscope.
Off-Target Analysis and Karyotyping
The off-target sites of the B2M CRISPR/Cas9 target were predicted by benchling (https://benchling.com/). Primers for the top three high-ranking off-target sites in whole genome and in coding sequences were designed accordingly. Those primer sequences were included in Table S1. Genomic DNA of B2MKO hP-iPSCs clones was isolated by DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instruction. Sequences of off-target sites were amplified by PCR, respectively, purified and analyzed by Sanger Sequencing (AITbiotech, Singapore). For karyotyping, confluent hP-iPSCs colonies were sent to the National University Hospital (NUH) referral laboratory (NRL, Singapore) for chromosome analysis.
Statistical Analysis
Data were collected as described above and summarized by Prism Version 7 software (GraphPad, CA, USA). Data were presented as mean (±SD) and data of cell cytotoxicity assay were analyzed by two-way analysis of variance, Tukey’s multiple comparisons test, and independent samples Student’s t-test (***P < 0.001; **P < 0.01; *P < 0.05). Representative histograms and graphs were chosen from independent repetition on the basis of the average values.
Results
Generation of B2MKO hP-iPSC clone by CRISPR/Cas9 With Double-Color Selection
To disrupt the B2M gene in human PBMC-derived iPSCs, we first target B2M by CRISPR/Cas9 with the help of color selection. hP-iPSCs were co-transfected with a CRISPR/Cas9 plasmid pX260 to target the B2M gene and a donor plasmid containing EGFP and a neomycin-resistant gene flanked by B2M homologous sequences (Fig. 1A). Transfected cells were selected by geneticin for 2 weeks and sorted by GFP positive expression as single cells. A total of 18 single-cell clones were collected and expanded for further analysis. All the 18 single clones were confirmed with the integration of the EGFP selection marker into the B2M site (Fig. S1A), but they still bore normal B2M expression. Sequencing analysis showed that they each had an intact wild-type allele of B2M (Fig. S1B), which indicated that these 18 clones were only monoallelic knockout.

Generation of B2M knockout hP-iPSCs with CRISPR/Cas9 and double-color selection. (A) Schematic of a CRISPR/Cas9 system targeting B2M EX1 with two selection donor templates for homologous recombination. The system was used for genetic modification in hP-iPSCs by electroporation. Arrows showed the binding sites of PCR primers for genotyping. (B) Representative images of B2M biallelic knockout hP-iPSC single-cell clone. The scale bar was 200 μm, and the exposure time was 1 s for fluorescence. (C) PCR analysis of WT and HDR alleles of the B2M gene from B2M monoallelic and biallelic knockout hP-iPSC single-cell clones. WT hP-iPSCs were included as a control. (D) Western blot analysis of B2M expression in B2M monoallelic and biallelic knockout hP-iPSC single-cell clones. Sample cells were treated by interferon-γ for 48 h before analysis. WT clone was used as a control, and β-actin was detected as housekeeping expression. (E) Representative flow cytometry diagrams of surface B2M and HLA-A, B, C expression on WT, B2M monoallelic and biallelic knockout hP-iPSC single-cell clones. CRISPR/Cas9: clustered regulatory interspaced short palindromic repeat-associated Cas9 endonuclease; EX1: exon 1; HDR: homology-direct recombination; HLA: human leukocyte antigen; hP-iPSCs: human peripheral blood mononuclear cell-derived induced pluripotent stem cells; PCR: polymerase chain reaction; WT: wild type.
To disrupt the remaining wild-type B2M allele, we transfected one of the monoallelic knockout clone #18 with B2M targeting CRISPR/Cas9 plasmid again but accompanied with a donor containing mCherry and Hygromycin selection marker (Fig. 1A). Similarly, transfected cells underwent Hygromycin B selection first for 2 weeks and then cell sorting which was based on EGFP and mCherry two colors. A total of six single-cell clones were collected and expanded showing positive for both selection markers (Fig. 1B). Besides, all the six clones were confirmed with site-specific integration of both markers in the B2M site, and no wild-type B2M allele could be detected by PCR (Fig. 1C). This suggests that all the two alleles of B2M were knocked out in the six double-color hP-iPSC clones. As expected, B2M expression could not be detected on those biallelic knockout clones by Western blotting, even when cells were treated by IFN-γ for 48 h (Fig. 1D and Fig. S1C). Monoallelic knockout clone, however, maintained potent B2M expression when compared with wild-type clone. This finding was also supported by flow cytometry assay showing the surface expression of B2M, while only biallelic knockout ones were negative for B2M expression (Fig. 1E and Fig. S1D). In addition, biallelic knockout clones were also negative for HLA class I (Fig. 1E and Fig. S1D). As B2M is the key component of the HLA class I complex, disruption of B2M could block HLA class I expression and substantially reduce the immunogenicity. Therefore, we have achieved the B2MKO hP-iPSCs with double-color selection.
One-Step Generation of B2MKO hP-iPSC Clone by CRISPR/Cas9 Without Color Selection
Although the color selection approach to facilitate CRISPR/cas9-based iPSC genomic editing is well established, the entire process could be time-consuming, less efficient, and lead to off-target cleavages 27 . We indeed observed that the cells with fluorescence would interfere with subsequent assays using relevant fluorescent reagents. Potential integration of selection marker into the genomic region besides B2M was also an arising concern, while southern blotting has indicated non-B2M site integration of the EGFP marker in both monoallelic and biallelic knockout cells (Fig. S1E). To overcome the above issues, we further developed a method to knockout B2M on hP-iPSCs without color selection.
We observed that, after the transfection of CRISPR/Cas9 plasmid, some cells were already B2M negative, but they, however, could not be selected. Therefore, the plasmid pX459 was introduced as a transient expression of Cas9 which could be selected by puromycin 24 . hP-iPSCs were transfected with pX459 plasmids targeting B2M by electroporation (Fig. S2A). When transfected cells were settled down, they were subsequently selected by puromycin for only 24 h. Within the survival cells, a large amount of B2M negative cells could be observed by flow cytometry assay (Fig. S2B). These survival cells were seeded as single cells, and 12 single-cell clones were successfully expanded. Among the 12 clones, five of them were tested as B2M negative by flow cytometry. B2MKO clone #3 and #8 were randomly selected for subsequent analysis. Both clones were negative for B2M and HLA class I molecules (Fig. 2A). By genotyping, we found either deletion or insertion in the B2M targeting site in the two B2MKO clones (Fig. 2B). Such a specific mutation, which was induced by CRISPR/Cas9 mediated cleavage, caused a frameshift of B2M translational reading frame and brought an early stop codon to terminate the B2M expression. The accuracy of this CRISPR/Cas9-mediated B2MKO was also examined by sequencing high-risk off-target sites. Only one off-target was spotted in clone #3, which did not affect the reading frame, while no observable off-target was found in clone #8, indicating the precision of the gene editing (Fig. S2C). In addition, karyotyping also showed the normal status of the chromosome in the B2MKO clone #8 (Fig. S2D). This B2MKO clone was used in subsequent analysis. Overall, these findings indicate that we knocked out the B2M gene on hP-iPSCs precisely without introducing any selection marker or random genomic damage.

One-step generation of B2MKO hP-iPSCs by CRISPR/Cas9 technology.
Maintenance of Pluripotency on B2MKO hP-iPSCs
The pluripotency of B2MKO iPSCs was validated. RT-PCR results showed equivalent expression of Oct4, Sox2, and Nanog in wild-type and B2MKO iPSCs (Fig. 3A). Monoallelic and biallelic B2MKO hP-iPSCs with color selection markers also had comparable expression of pluripotent markers (Fig. S3A). Both wild-type and B2MKO iPSCs were induced to form EBs (Fig. 3B and Fig. S3B), from which three germ layer differentiation could be observed. All three germ layer differentiation markers were detected in B2MKO iPSC-derived EBs as well as in wild-type iPSC-derived EBs by RT-PCR (Fig. 3C and Fig. S3C) and immunostaining (Fig. 3D), which suggested the intact differentiation capacity of B2MKO iPSCs.

Pluripotency of B2MKO hP-iPSCs. (A) RT-PCR analysis of pluripotent marker Oct4, Sox2, and Nanog expression in WT and B2MKO hP-iPSC single-cell clones. The expression of β-actin was included as a housekeeping control. (B) Representative images of EBs derived from WT and B2MKO hP-iPSC single-cell clones. The scale bar was 200 μm. (C) RT-PCR analysis of markers’ expression of three germ layers in WT and B2MKO hP-iPSC-derived EBs. Pax6 was detected for ectoderm; MHC-α was detected for mesoderm, and AFP was detected for endoderm. The expression of β-actin was included as a housekeeping control. (D) Immunostaining of three germ layer markers in WT and B2MKO hP-iPSC-derived EBs. Beta-III-tubulin (TUJ1) was stained for ectoderm in blue color; SMA was stained for mesoderm in red color, and AFP was stained for endoderm in green color. The scale bar was 500 μm. AFP: alpha-fetoprotein; B2MKO: B2M knockout; EB: embryoid bodies; hP-iPSCs: human peripheral blood mononuclear cell-derived induced pluripotent stem cells; SMA: smooth muscle actin; RT-PCR: reverse transcription-polymerase chain reaction; WT: wild type.
Generation of MSCs from B2MKO hP-iPSCs
Many studies have unveiled the feasibility of deriving MSCs from human embryonic stem cells (hESCs) or iPSCs 13,21,28,29 . In this study, we adapted a method of generating MSCs from B2MKO hP-iPSCs directly 21 . The MSCs were successfully derived from B2MKO iPSCs as well as wild-type iPSCs. Both showed the phenotype of fibroblast-like cells and were named iMSCs (Fig. 4A). Flow cytometry assay confirmed negative for B2M and HLA class I on iMSCs derived from B2MKO iPSCs (Fig. 4B). Further phenotyping supported that those B2MKO cells maintained the characteristics of MSCs as they were negative for CD14, CD24, CD34, CD45, and HLA-DR but positive for CD29, CD44, CD73, CD90, CD105, and CD166 (Fig. 4C). The multipotency of B2MKO iPSC-derived iMSCs was analyzed by inducing further differentiation of these iMSCs into adipocytes, osteocytes, and chondrocytes. Adipogenesis was confirmed by positive staining of lipid content in differentiated cells with Oil Red O (Fig. 5A). Robust osteogenesis was verified by positive red staining of calcific deposition with Alizarin Red S (Fig. 5B). In addition, chondrogenesis was achieved with the formation of chondrocyte aggregates and blue staining of acidic polysaccharides (Fig. 5C). RT-PCR results also showed upregulation of relevant markers in differentiated cells supporting the differentiation capacity of B2MKO iPSC-derived iMSCs (Fig. 5D–F). Thus, functional iMSCs could be generated from B2MKO hP-iPSCs.

Generation of mesenchymal stromal cells from B2MKO hP-iPSCs. (A) Representative images of WT and B2MKO hP-iPSC-derived MSCs. The scale bar was 200 μm. (B) Representative flow cytometry diagrams of surface B2M and HLA-A, B, C expression on WT and B2MKO iMSCs. (C) Phenotyping of WT and B2MKO iMSCs by surface markers. The expression of MSC negative markers (CD14, CD24, CD34, CD45, and HLA-DR) and MSC positive markers (CD29, CD44, CD73, CD90, CD105, and CD166) was shown as representative flow cytometry diagrams. B2MKO: B2M knockout; HLA: human leukocyte antigen; hP-iPSCs: human peripheral blood mononuclear cells-derived induced pluripotent stem cells; MSCs: mesenchymal stromal cells; WT: wild type.

Multipotency of B2MKO hP-iPSC-derived MSCs. (A) Adipogenesis, (B) osteogenesis, and (C) chondrogenesis of WT and B2MKO iMSCs. Lipid content was stained in red by Oil Red O for adipocytes; calcific deposition was stained in red by Alizarin Red S for osteocytes, and acidic polysaccharides were stained in blue by Alcian blue 8GX for chondrocytes. The scale bar for all the images was 80 μm. Expression of specific markers was detected by RT-PCR for both WT and B2MKO iMSCs and differentiated cells. (D) LPL and PPAR-γ were detected for adipogenesis. (E) Bone relevant ALP and OCN were detected for osteogenesis. (F) ACAN and COL2a were detected for chondrogenesis. ACAN: aggrecan; ALP: alkaline phosphatase; B2MKO: B2M knockout; COL2a: collagen type II alpha 1; hP-iPSCs: human peripheral blood mononuclear cell-derived induced pluripotent stem cells; LPL: lipoprotein lipase; MSC: mesenchymal stromal cells; OCN: osteocalcin; PPAR-γ: peroxisome proliferator-activated receptor-gamma; RT-PCR: reverse transcription-polymerase chain reaction; WT: wild type.
Effects of iMSCs on T Cell Proliferation
In view of the potent immunosuppressive effects of MSCs, we performed mixed lymphocyte reaction (MLR) assays to examine the effects of iMSCs on T cell proliferation. Allogeneic T cells from PBMCs were activated by anti-CD3 antibodies (OKT-3) and mixed with wild-type or B2MKO iMSCs, followed by tracking the proliferation of the T cells by Far Red staining and analyzing with flow cytometry. The histograms of Far Red staining in activated T cells with or without iMSCs co-culture were displayed in Fig. 6A. As shown, only 3 days after activation, in the group without iMSCs, proliferating T cells had contributed to 68% of the total population with four generations of Far Red dye dilution. As expected, when co-culturing with wild-type iMSCs, the population of the proliferating T cells was reduced to 58%, displaying reduced, three generations of dye dilution by day 3. To our surprise, when B2MKO iMSCs were introduced, the percentage of the proliferating T cells was further reduced to 32%, with only two generations of dye dilution by day 3. Such immunosuppression on allogeneic T cell proliferation by B2MKO iMSCs was consistent among different donors, as summarized in Fig. 6B. While our results were consistent with previous studies 14,15 and demonstrated the inhibition of T cell proliferation by iMSCs, an even stronger immunosuppression provided by B2MKO iMSCs as comparing with wild type iMSCs is a new finding of possible clinical importance.

Immunosuppressive property of B2MKO hP-iPSC-derived MSCs. (A) Tracking proliferation of stimulated PBMCs in the presence or absence of iMSCs by Far Red staining. iMSCs were introduced in a PBMCs/iMSCs ratio of 2:1. Allogeneic PBMCs were stimulated by 50 ng/ml OKT-3 and the dilution of Far Red staining was gated. Representative set of flow cytometry histograms from three individual donors was shown. (B) Allogeneic PBMCs proliferation was summarized according to the percentage of relative proliferation. The dilution of Far Red in stimulated PBMCs without iMSCs was normalized as 100% in relative proliferation, while the dilution of Far Red in PBMCs without OKT-3 was 0% in relative proliferation. Bars show the mean ± SD of relative proliferation in three independent experiments with individual donors. **P < 0.01; *P < 0.05. B2MKO: B2M knockout; hP-iPSCs: human peripheral blood mononuclear cell-derived induced pluripotent stem cells; MSC: mesenchymal stromal cells; PMBC: peripheral blood mononuclear cell; RT-PCR: reverse transcription-polymerase chain reaction.
Hypoimmunogenicity of B2MKO hP-iPSC-Derived MSCs
To examine the immunogenicity of hP-iPSC-derived MSCs, we first primed allogeneic human PBMCs with wild-type iMSCs. Human PBMCs were co-cultured with wild-type iMSCs directly and restimulated by fresh iMSCs every week. Primed PBMCs were phenotyped by flow cytometry assay to delve into the specific population. Most primed donors would end up with a major CD3+CD56− T cell population with dominant CD8+ T cells (Fig. 7A). These primed PBMCs population indicated that alloreactive cytotoxic T lymphocytes had been stimulated by iMSCs, although MSCs or iMSCs were reported with immunosuppressive characteristics 14 –16 . When we challenged both wild-type and B2MKO iMSCs with those primed PBMCs in a cell cytotoxicity assay, to our expectation, wild-type iMSCs were preferentially killed by primed PBMCs but B2MKO iMSCs escaped from the killing (Fig. 7B). The B2MKO iMSCs could survive due to the loss of HLA class I antigen presenting while wild-type iMSCs were eliminated by alloreactive T cells. For certain donors, a substantial amount of CD3−CD56+ NK cells could remain accompanied with alloreactive T cells after priming (Fig. 7C). When these cells were used as effectors in the cell cytotoxicity assay, B2MKO iMSCs could also be killed dose-dependently but their cell lysis was dramatically lower than the wild-type counterpart (Fig. 7D). To evaluate iMSCs’ susceptibility to NK cells, we challenged both wild-type and B2MKO iMSCs with primary NK cells. Slightly higher lysis of B2MKO iMSCs was observed comparing with lysis of wild-type iMSCs (Fig. 7E), which was expected according to the “missing-self” theory 30 . Hence, B2MKO hP-iPSC-derived MSCs showed much lower immunogenicity to allogeneic PBMCs but slightly higher susceptibility to NK lysis.
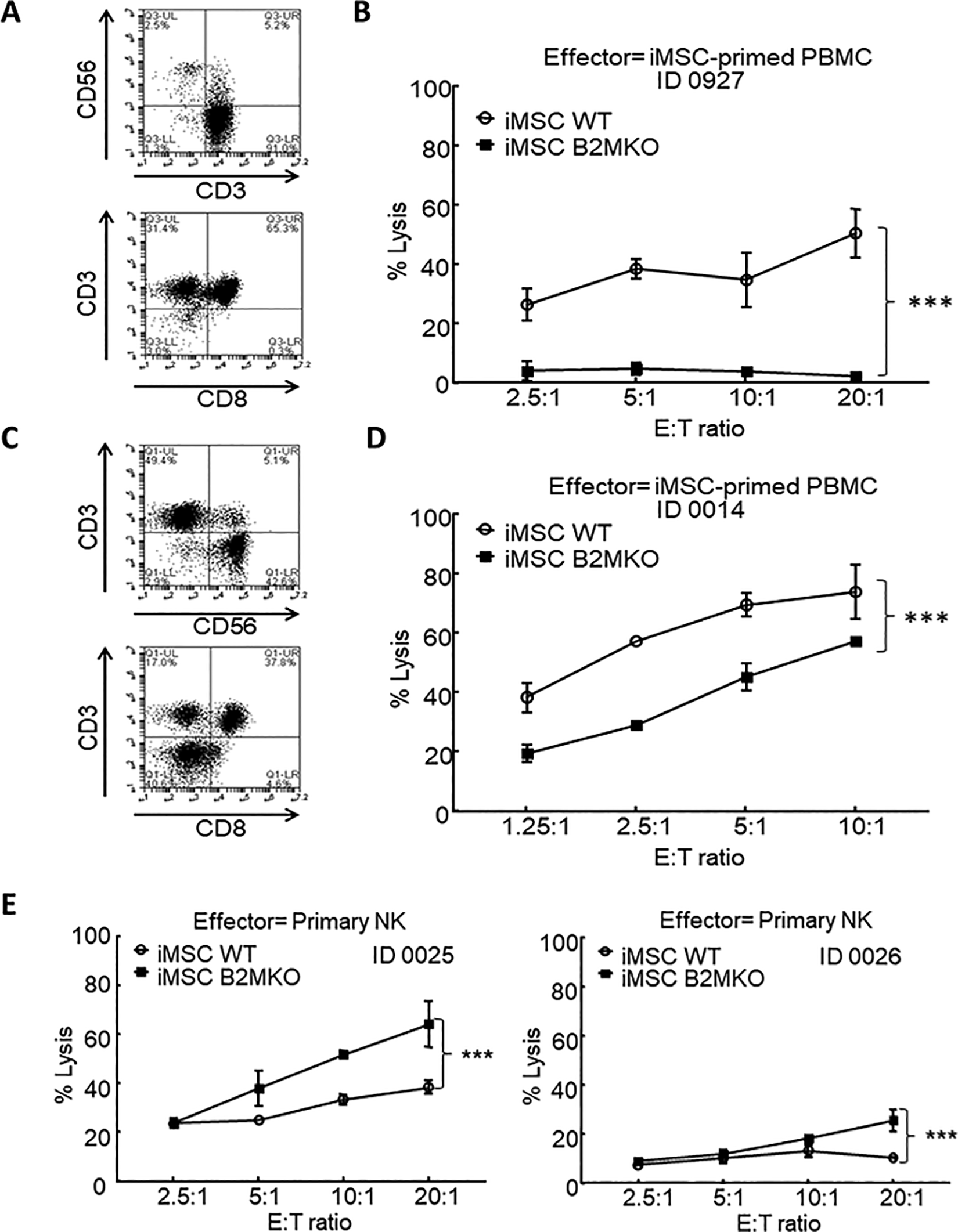
Hypoimmunogenicity of B2MKO hP-iPSC-derived mesenchymal stromal cells. (A, C) Phenotyping of iMSC-primed PBMCs was shown as flow cytometry diagrams for CD3, CD56, and CD8 expression. (B, D) Immunogenicity of both WT and B2MKO iMSCs when they were challenged by iMSC-primed PBMCs. The immunogenicity was examined with DELFIA EuTDA cytotoxicity assays (2 h Eu-ligand release) as target cells challenged by iMSC-primed PBMCs. (E) The susceptibility of WT and B2MKO iMSC to NK lysis. Primary NK cells were used as effectors to target WT and B2MKO iMSCs in DELFIA EuTDA cytotoxicity assays (2 h Eu-ligand release). For cell cytotoxicity assays, three independent assays from three individual donors were performed. Shown are percentage lysis of target cells at varying E:T ratios in one representative experiment (mean ± SD of triplicate samples). ***P < 0.001. B2MKO: B2M knockout; hP-iPSCs, human peripheral blood mononuclear cell-derived induced pluripotent stem cells; MSC: mesenchymal stromal cells; NK: natural killer; PMBC: peripheral blood mononuclear cell; WT: wild type.
Discussion
MSCs have been extensively characterized and broadly applied in adoptive cell transfer. To standardize the definition of this kind of multipotent stem cells, a minimum criterion for MSCs has been proposed by the International Society for Cellular Therapy in 2006 31 , saying that MSCs are (1) able to adhere to plastic in culture in vitro; (2) positive for markers such as CD105, CD73, and CD90, while negative for markers such as CD34, CD45, CD14, CD19, and HLA-DR; and (3) able to differentiate into the three lineages of adipocytes, osteocytes, and chondrocytes in vitro. MSCs could be derived from iPSCs through the formation of EBs 28,32 or neural crest 29 . In some protocols, a cell sorting based on the positive expression of MSC relevant markers, like CD105 or CD73, was also involved. Comparing the above methods, some studies displayed a more straightforward approach to generate MSCs from human iPSCs by simple culture medium substitution and subculture 21,28,32,33 .
In this study, we adapted the straightforward method to directly differentiate iPSCs into MSCs by changing the culture medium. As multipotent stem cells, MSCs were reported to differentiate into adipocytes, osteocytes, and chondrocytes. All three lineages of differentiation were confirmed with our wild-type and B2MKO iMSCs, among which the commitment into osteoblast and chondrocytes was favored. This was consistent with a previous study showing that iPSC-derived MSCs would be prone to differentiate into osteocytes and chondrocytes but not adipocytes 13 . Some lineage-specific markers were also examined. To our surprise, tissue-specific markers like peroxisome proliferator-activated receptor gamma, osteocalcin, aggrecan, and collagen type II alpha 1 were already detectable in iMSCs (Fig. 5D–F), indicating a variable transcriptional status of iMSCs that were likely to differentiate.
In addition to the tri-lineage potential (bone, cartilage, fat), MSCs have also been considered as vascular stem cells given their vascular or perivascular localization, although in vitro differentiation of MSCs to generate endothelial cells, pericytes, and vascular smooth muscle cells has yet to be demonstrated 34 . Nevertheless, promoting angiogenesis as an important role that MSCs can play is well documented 34,35 . This function could be beneficial in terms of regenerative medicine. Recent work has demonstrated that human iPSC-MSCs provided more potent angiogenic effects than human embryonic stem cell-derived cardiomyocytes in an animal transplantation study 35 . However, MSCs are one of the pivotal types of cells composing the tumor microenvironment and can mediate tumorigenesis, angiogenesis, and metastasis 36 . The dual role of MSCs in regenerative medicine and cancer progression calls for more extensive research that can clearly identify MSC functions at the physiological and molecular levels.
MSCs are considered as immune-privileged cells and were widely applied in allogeneic scenarios. Allogeneic MSCs were even used to alleviate GvHDs after bone marrow transplantation. Although no adverse effect was reported in the clinical trials of allogeneic MSCs, their immunogenicity was still a concern because induced allogeneic immune memory was observed in previous animal studies 16 . In this study, we confirmed the potent HLA class I expression on our wild-type hP-iPSC-derived MSCs, which could trigger the allogeneic rejection. We were able to induce the alloreactive PBMCs by priming allogeneic PBMCs with wild-type iMSCs. The allogeneic rejection was demonstrated with cell cytotoxicity assay in vitro, showing the cytotoxicity of alloreactive PBMCs against wild-type iMSCs but not B2MKO iMSCs. This in vitro study revealed the privilege of B2MKO iMSCs in allogeneic T cell-mediated rejection. Although wild-type iMSCs should be able to suppress the growth of alloreactive T cells, such immunosuppression might be eventually overcome by the allogeneic response against their HLA class I expression. In this case, wild-type iMSCs could be eliminated by alloreactive T cells before those iMSCs executed their immunosuppressive function. Based on the above deduction, B2MKO iMSCs may provide a prolonged therapeutic effect with their ability to dodge the allogeneic rejection.
Interestingly, we observed that B2MKO iMSCs provided an enhanced immunosuppression to T cell proliferation in an MSC/MLR assay when compared with wild-type iMSCs. Such immunomodulation mediated by MSCs could be due to a direct cell-to-cell ligation, a paracrine signaling of inhibitory factors, or even the inhibition of caspase cleavage 37 –39 . As reported previously, the Rap1-modulated nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling plays a key role in MSC immunomodulation 40,41 . However, it has already been demonstrated that B2M stimulates NF-κB pathways, at least in cancer cells 42 , suggesting that the Rap1/NF-κB signaling may not be a key player in mediating the enhanced immunosuppression by B2MKO iMSCs observed in the present study. On the other hand, B2M free, soluble HLA Class I molecules have been shown to inhibit the activity of activated T lymphocytes and even induce apoptosis of T cells 43,44 . Along the line, Masuda et al, have reported that adipose tissue-derived MSCs isolated from B2M-deficient mice display a potent capacity to suppress IFN-γ production 45 . It would be worth further investigation in a future study whether the actions of soluble HLA Class I molecules are the underlying mechanism of the enhanced inhibition to T cell proliferation associated with B2MKO iMSCs.
According to the “missing-self” theory, NK cells were to be activated and preferentially lyse the HLA class I-negative cells 30 . This theory was supported in our study showing increased NK susceptibility of B2MKO iMSCs. Compared with the T cell-mediated adaptive immunity, such NK dominant innate immunity contributed less to the allogeneic rejection. However, this could be favorably applied to eliminate B2MKO iMSCs as MSCs were reported to promote tumor angiogenesis and metastasis 35 . NK susceptibility may reduce the risk of tumorigenesis when B2MKO iMSCs start to display cancer progression promoting effects. To reduce the NK susceptibility of B2MKO cells, as reported in Gornalusse et al 46 and Zhao et al’s studies 47 , nonclassical HLA complex, such as HLA-G and HLA-E, could be introduced to inhibit the NK function. HLA-G is the ligand of inhibitory receptor KIR2DL4, while HLA-E is the ligand of inhibitory receptor NKG2A/CD94 complex. Both of them could suppress the function of allogeneic NK cells without triggering T cell-mediated allogeneic responses. However, such modification is a controversy that when HLA-E and HLA-G protect graft cells from NK lysis, they could broadly suppress the NK function in the recipient and impair inherent innate immunity. Further studies should be conducted to investigate the functionality of non-classical HLA modified cells after adoptive cell transfer in vivo.
The hiPSCs were first achieved by Shinya Yamanaka’s lab in 2007 48 . hiPSCs now hold great promise for the clinical application in stem cell-based therapy, while clinical studies of hiPSC derivatives were limited due to the heterogeneity of iPSCs from different individuals and the inconvenience in the generation of personalized iPSCs. Many previous studies have attempted to generate hypoimmunogenic stem cells by disrupting B2M expression. As reported, those pluripotent stem cells maintained their characteristics after the B2M disruption, which validated the feasibility of B2MKO in our hP-iPSCs. However, the efficiency of B2MKO remained to be enhanced. Riolobos et al 49 first disrupted the B2M gene in human ESCs by AAV mediated gene targeting, but trace of B2M expression was spotted in their lines in a later publication showing a separate start codon in the disruptive homologous recombination. They next improved the targeting method to eventually collect B2M biallelic knockout clones by repeated subcloning, however, with abnormal karyotypes 46 . With the development of gene editing tools, transcription activator-like effector nucleases (TALENs) and CRISPR/Cas9 system were introduced to benefit B2M disruption in human pluripotent stem cells. Lu et al 50 used TALENs to target B2M in hESCs, while Sato et al 51 achieved similar B2MKO by CRISPR/Cas9 in human iPSCs. Both B2MKO pluripotent stem cell clones were generated without the integration of exogenous genes.
Here, we knocked out B2M in human PBMC-derived iPSCs with two different methods. We first followed the gene targeting using double-color homologous recombination and CRISPR/Cas9. B2MKO hP-iPSC clones were finally achieved after two rounds of color selection and single-cell cloning. Although double-color selection was a visible method, such knockout efficiency was relatively low and the knockout clones might also bear the risk of random integration. To improve the B2MKO efficiency and secure the genome stability, we further generated B2MKO hP-iPSCs in a way similar to Sato et al’s publication 51 . In this method, we only transiently selected the expression of Cas9 effector endonuclease. Yet, the knockout efficiency was dramatically increased to around 50%. Such one-step knockout method created site-specific mutation in B2M locus, which would not affect any other gene locus or introduce any exogenous sequence into the genome. CRISPR/Cas9 was reported with possibility of off-target in previous studies 52,53 . It was also confirmed by our practice as one off-target induced mutation was spotted in B2MKO#3 single-cell clone. To minimize such off-target effect, the guide RNA sequences could be optimized to reduce the chance of mismatched binding and a high-fidelity version of Cas9 could be utilized to prevent unwanted DNA cleavage 52 . Besides, a more precise CRISPR system could also be adopted to improve the specificity of such gene editing. With the selection and examination of single-cell clones, genome integrity can be controlled in gene editing of iPSCs.
Conclusion
In conclusion, this study has developed a new method to generate B2MKO hP-IPSC-derived MSCs. We first knocked out B2M gene by CRISPR/Cas9 with or without color selection and achieved the HLA class I-negative iPSCs. The B2MKO hP-iPSCs were verified with intact pluripotency and genome stability. Second, based on this B2MKO hP-iPSC clone, we derived B2MKO MSCs directly. The B2MKO iMSCs were characterized by the expression of proper markers, multipotency to differentiate, and immunosuppressive property. Finally, these B2MKO iMSCs showed lower immunogenicity to allogeneic PBMCs with a potential for “off the shelf” cell resources.
Supplemental Material
Supplemental Material, Generation_of_mesenchymal_stromal_cells_with_low_immunogenicity_from_human_PBMC - Generation of Mesenchymal Stromal Cells with Low Immunogenicity from Human PBMC-Derived β2 Microglobulin Knockout Induced Pluripotent Stem Cells
Supplemental Material, Generation_of_mesenchymal_stromal_cells_with_low_immunogenicity_from_human_PBMC for Generation of Mesenchymal Stromal Cells with Low Immunogenicity from Human PBMC-Derived β2 Microglobulin Knockout Induced Pluripotent Stem Cells by Shijun Zha, Johan Chin-Kang Tay, Sumin Zhu, Zhendong Li, Zhicheng Du and Shu Wang in Cell Transplantation
Footnotes
Ethical Approval
This study was approved by our institutional review board.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Disclosure Statement
The authors SZ and SW have filed a patent application related to the iPSC-iMSC technology and could potentially receive licensing royalties in future.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Singapore Ministry of Health’s National Medical Research Council (NMRC/CIRG/1406/2014). The authors declare that there is no conflict of interest.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.