
Abstract
The aim of our study is to determine whether insulin-producing cells (IPCs) differentiated from adipose-tissue-derived stem cells (ADSCs) can be cryopreserved. Human ADSCs were differentiated into IPCs using our two-step protocol encompassing a three-dimensional culture and xenoantigen-free method. Thereafter, IPCs were frozen using three different methods. First, IPCs were immediately frozen at −80°C (−80°C group). Second, IPCs were initially placed into a Bicell freezing container before freezing at −80°C (BICELL group). Third, a vitrification method for oocytes and embryos was used (CRYOTOP group). Cell counting kit-8 (CCK-8) assay showed that cell viability was decreased in all groups after cryopreservation (P < 0.01). Corroboratively, the amount of adenosine triphosphate was markedly decreased after cryopreservation in all groups (P < 0.01). Immunofluorescence staining showed a reduced positive staining area for insulin in all cryopreservation groups. Furthermore, 4′,6-diamidino-2-phenylindole and merged immunofluorescence images showed that cryopreserved cells appeared to be randomly reduced in the −80°C group and CRYOTOP group, while only the central region was visibly reduced in the BICELL group. Using immunohistochemical staining, IPCs after cryopreservation were shown to be positive for cleaved caspase-3 antibody in all groups. Finally, insulin secretion following glucose stimulation was significantly reduced in IPCs from all groups after cryopreservation (P < 0.01). In conclusion, IPCs may be too fragile for cryopreservation with accomplished methods and further investigations for a suitable preservation method are required.
Introduction
Diabetes mellitus affects over 382 million people worldwide and this number is expected to rise to 592 million by 2035 1 . Pancreatic transplantation and pancreatic islet transplantation are considered options for the cure of type 1 diabetes mellitus (T1DM). However, the Edmonton protocol 2 (established in 2000 for islet transplantation only) showed that the insulin-independent rate at 5 years after islet transplantation was less than 10%, even though islet transplantation is still a minimally invasive option for T1DM 3 . In Europe and North America, where there are relatively abundant donor sources, multidonor–one recipient transplantation is possible for such circumstances. However, in some countries such as Japan, it is difficult to secure sufficient islets owing to a severe donor shortage. Thus, it is important to investigate new cell sources as a fundamental solution. One possibility is the use of regenerative medicine as a new cell source, i.e., induced pluripotent stem (iPS) cells, embryonic stem (ES) cells, and mesenchymal stem cells (MSCs). Nonetheless, various problems have been noted, such as the risk of genetic damage and alloreaction with iPS cells and ethical problems of ES cells. Therefore, it may take some time before there is clinical application of these two cell sources. Alternatively, MSCs can solve these problems, particularly autotransplantation of MSCs using regenerative medicine. Among MSCs, adipose-tissue-derived stem cells (ADSCs) can be collected from a patient’s own fat tissues in a minimally invasive and easy approach compared with other MSCs. Correspondingly, we have investigated differentiation of ADSCs to insulin-producing cells (IPCs), which are monoclonal and have pancreatic β cell-like functions. Appropriately, we have generated IPCs from ADSCs using our established, easy, and rapid two-step protocol, which encompasses a three-dimensional culture using recombinant peptide (RCP) micro-pieces (μ-piece) and a xenoantigen-free method 4 –8 . Indeed, our established protocol can be considered to have reached the nonclinical “proof of concept” stage; therefore, when considering the tests required by government regulations or the need to transport IPCs to other institutes for clinical application, it is important to determine how IPCs can be properly preserved. However, it is still unclear whether IPCs generated by our strategy can be cryopreserved, even though there have been several reports on pancreatic islet preservation methods 9 –14 . If IPC cryopreservation is possible, the issue of time-to-transplant can be minimized and shipping to remote institutes may be feasible. Furthermore, it can lead to the mechanism of the reaction in the cryopreservation of IPCs and improve transplantation results. Here, we investigate the possibility of IPC cryopreservation using IPCs generated by our established protocol.
Materials and Methods
Cell Preparation
STEMPRO™ Human Adipose-Derived Stem Cells (Thermo Fisher Scientific, Inc., Waltham, MA, USA) were used for this project. ADSCs were passaged using an ADSC basal medium mixture of MesenPRO™ RS (Thermo Fisher Scientific, Inc.) and GlutaMAX™-Ⅰ (Thermo Fisher Scientific, Inc.). After sufficient passaging, ADSCs were counted. Next, 2.0 × 106 ADSCs and 0.2 mg/ml RCP μ-pieces (Fujifilm Corporation, Tokyo, Japan) were mixed evenly and injected into all wells of a Nunclon Sphera 96U Bottom Plate (Thermo Fisher Scientific, Inc.) as a three-dimensional culture. IPCs were generated using our rapid two-step, three-dimensional culture and xenoantigen-free protocol 8 . Briefly, ADSCs were cultured using Step-1 (from day 0 to day 7) IPC differentiation medium containing Dulbecco’s modified Eagle’s medium/F12 (Thermo Fisher Scientific, Inc.), 1% recombinant human albumin (Fujifilm Wako Pure Chemical Corporation, Osaka, Japan), 10 nM exendin-4 (Sigma–Aldrich Co., St. Louis, MO, USA), 1% N2 supplement (Thermo Fisher Scientific, Inc.), 1% B27 supplement (Thermo Fisher Scientific, Inc.), and 50 ng/ml recombinant human activin-A (PeproTech Inc., Rocky Hill, NJ, USA). Step-2 medium (from day 8 to day 21) was prepared the same way as Step-1 medium with the addition of 50 ng/ml recombinant human hepatocyte growth factor (PeproTech Inc.), 0.016% valproic acid (Fujifilm Wako Pure Chemical Corporation), and 10 mM nicotinamide (Sigma–Aldrich Co.) in culture conditions of 37 °C and 5% CO2. The culture medium was changed and supernatant collected every 2 days.
Cryopreservation
After completion of the culture, IPCs were washed two times with Hank’s balanced salt solution. Then IPCs were cryopreserved in three different ways. First, 5.0 × 105 IPCs were aliquoted into a 1.0 mm cryotube (Thermo Fisher Scientific, Inc.) together with 1.0 ml preservative solution, COS banker Ⅱ (Cosmo Bio Co., Ltd., Tokyo, Japan), and frozen immediately at −80 °C (−80 °C group). The second method was to aliquot 5.0 × 105 IPCs together with 1.0 ml of the same preservative solution into 1.0 mm cryotubes. The cryotube was then placed into a freezing container (Bicell; Nihon Freezer Co, Ltd., Tokyo, Japan) before freezing at −80 °C (BICELL group). The last approach incorporated a vitrification method for oocytes and embryos (Cryotop safety kit; Kitazato Corporation, Tokyo, Japan) (CRYOTOP group). After cryopreservation for 48 h, IPCs were rapidly thawed in a water bath at 37 °C or using the Cryotop safety kit thawing protocol. ADSCs or a human pancreas cancer cell line (Panc-1) was used as a control.
CCK-8 Assay
Cell viability of each sample was determined using the CCK-8 (Dojindo Molecular Technologies, Inc., Tokyo, Japan) according to the manufacturer’s protocol. Briefly, IPCs or ADSCs were preincubated in 96-well plates at 37 °C and 5% CO2. Next, 10 µl CCK-8 solution was added to each well, followed by color development for 4 h. Sample plates were used to measure optical absorbance at 450 nm. Optical absorbance was measured using a SpectraMax i3 (Molecular Devices, LLC, San Jose, CA, USA) and SoftMax Pro 7 (Molecular Devices, LLC). Cell viability before cryopreservation was denoted as 100% and compared with cell viability after cryopreservation.
Adenosine Triphosphate Assay
The adenosine triphosphate (ATP) concentration in each sample was measured using the CellTiter-Glo® 3D Cell Viability Assay (Promega Corporation, Madison, WI, USA), according to the manufacturer’s protocol. Briefly, CellTiter-Glo® 3D Reagent was added to an equal volume of cell culture medium present in each well and the contents mixed vigorously for 5 min to induce cell lysis. The plate was incubated at room temperature for an additional 25 min to stabilize the luminescent signal before it was recorded. Luminescence intensity was measured using SpectraMax i3 and SoftMax Pro 7. ATP values were compared before and after cryopreservation.
Immunohistochemical Staining
IPC samples were formalin fixed overnight and paraffin embedded in iPGell (Genostaff Co., Ltd., Tokyo, Japan). Six-micrometer-sliced sections of IPC samples were dewaxed, deparaffinized in xylene (Fujifilm Wako Pure Chemical Corporation), and rehydrated through a series of graded alcohols. Endogenous peroxidases were blocked with 0.3% hydrogen peroxidase (Fujifilm Wako Pure Chemical Corporation) for 20 min. For antigen retrieval, sections were boiled with ethylenediaminetetraacetic acid buffer (pH 6.0) in a pressure cooker for 15 min (1,400 W) and 10 min (700 W). Sections were then incubated in Protein Block Serum-Free (Agilent Technologies, Inc., Santa Clara, CA, USA) for 10 min to prevent nonspecific antigen binding. Sections were incubated with primary antibody at a dilution of 1:100 in phosphate-buffered saline (PBS) overnight at 4 °C. The primary antibody used was cleaved caspase-3 antibody (#9661; Cell Signaling Technology, Inc., Danvers, MA, USA). Sections were incubated in secondary antibody (EnVision Dual Link System-HRP; Agilent Technologies, Inc.) for 1 h at room temperature. Sections were developed using diaminobenzidine (Fujifilm Wako Pure Chemical Corporation) and counterstained with Mayer’s hematoxylin (Muto Pure Chemicals, Co., Ltd., Tokyo, Japan). Sections were dehydrated in graded alcohols and covered with coverslips.
Immunofluorescence Staining
Blocking of endogenous peroxidases and antigen retrieval were performed as described above. To prevent nonspecific antigen binding, sections were incubated in 3% bovine serum albumin (BSA) for 1 h. Thereafter, sections were incubated with primary antibodies, namely, anti-insulin (1:1,000, ab6995; Abcam plc., Cambridge, UK) for 1 h at room temperature. After washing with PBS three times, sections were then incubated with a secondary antibody, Alexa Fluor 594 goat anti-mouse immunoglobulin G (1:500, A-11005; Thermo Fisher Scientific, Inc.), for 1 h in the dark. Following further PBS washes, 4′,6-diamidino-2-phenylindole (DAPI) (P-36931; Thermo Fisher Scientific, Inc.) was applied to the slides.
Glucose-stimulated Insulin Secretion Test
The glucose-stimulated insulin secretion test was performed according to previous reports 15,16 . Briefly, 3.3 × 105 IPCs were preincubated for 2 h in 2 ml RPMI medium supplemented with 0.5% BSA (Sigma–Aldrich Co.) and 2.2 mM glucose (Fujifilm Wako Pure Chemical Corporation). Thereafter, IPCs were stimulated in RPMI medium with the addition of various glucose concentrations (basal I: 2.2 mM; stimulation: 22 mM) for 1 h at 37 °C and 5% CO2. After stimulation, supernatants of each sample were collected and human insulin concentration was measured by enzyme-linked immunosorbent assay (ELISA) using the Human/Canine/Porcine Insulin Quantikine ELISA Kit (R&D Systems, Inc., Minneapolis, MN, USA). Optical absorbance in ELISA was measured using SpectraMax i3 and SoftMax Pro 7.
Dithizone Staining
Cultured cells were stained using dithizone solution. Dithizone solution contained 50 mg dithizone (Fujifilm Wako Pure Chemical Corporation) per 5 ml dimethyl sulfoxide (Fujifilm Wako Pure Chemical Corporation). After washing in PBS three times, IPCs were incubated in dithizone solution at 37 °C and 5% CO2. Stained samples were investigated using a multipurpose microscope BZ-X710 (Keyence Engineering Corporation, Osaka, Japan) and BZ-X Analyzer (Keyence Software Corporation).
Imaging Quantitative Analysis
Cell images were digitally analyzed by Image J (National Institute of Health, Bethesda, MD, USA) 17 . For analysis of dithizone staining, cell images were divided into three channels using the hue, saturation, and value (HSV) model, and filtered using a hue threshold of 7–20. Mean gray saturation values of three random points in a spheroid area were measured in filtered images as the stained intensity. For immunohistochemistry analysis, after dividing images into three channels, sample images were filtered using a hue threshold of 20–28. Mean gray saturation values of three random points in a spheroid area and the background were measured in filtered images. Staining intensity of immunohistochemistry was calculated as the ratio of the mean gray value in the spheroid area divided by the background.
Statistical Analysis
Data analysis was performed with statistical software (JMP software, version 13; SAS Campus Drive, Cary, NC, USA). Comparisons between two groups were performed by Mann–Whitney U test. In the figures, median ± standard deviation is provided. A value of P < 0.05 was used to indicate statistical significance.
Results
Decreased Dithizone Staining Intensity After Cryopreservation
After cryopreservation, the border of IPCs was less distinct and cell cluster structures appeared to be slightly collapsed compared with IPCs before cryopreservation (Fig. 1A, upper row). The appearance of dithizone staining in IPCs after cryopreservation was weaker than before cryopreservation (Fig. 1A, lower row). Further, Image J analysis of IPCs showed that the dithizone staining intensity was significantly decreased after cryopreservation in all groups (average staining intensity before cryopreservation: 239.3; −80 °C group: 211.6; BICELL group: 192.3; CRYOTOP group: 206.8; P < 0.05, Mann–Whitney U test, Fig. 1B).

Morphology of cryopreserved IPCs. (A) The borders of cryopreserved IPCs were faint (upper row). Dithizone staining of cryopreserved IPCs was weak (lower row). Representative images of three independent experiments are shown. Scale bar: 200 µm. (B) Image J analysis showed that dithizone staining intensity was significantly decreased by cryopreservation (P < 0.01, Mann–Whitney U test). IPC: insulin-producing cell.
Significantly Reduced Cell Viability After Cryopreservation
Compared with before freezing, absorbance in the CCK-8 assay was significantly decreased in all cryopreservation groups (average cell viability of −80 °C group: 13.3%; BICELL group: 6.3%; CRYOTOP group: 15.6%; P < 0.01, Mann–Whitney U test, Fig. 2). In contrast, the viability of ADSCs did not significantly change after cryopreservation.

CCK-8 assay. Cell viability determined by CCK-8 assay was significantly decreased in all cryopreservation groups (P < 0.01, Mann–Whitney U test). In comparison, cell viability did not change in ADSCs. ADSCs: adipose-tissue-derived stem cells; CCK-8: cell counting kit-8.
Decreased ATP Generated in Cryopreserved IPCs
Similarly, in the ATP assay, the amount of ATP concentration decreased significantly after cryopreservation (average ATP concentration before cryopreservation: 2.5 μM; −80 °C group: 0.4 μM; BICELL group: 0.1 μM; CRYOTOP group: 0.4 μM; P < 0.01, Mann–Whitney U test, Fig. 3A). Meanwhile, the amount of ATP of Panc-1 cells did not change statistically after cryopreservation (Fig. 3B).

ATP assay. (A) ATP production was significantly decreased in all cryopreservation groups (P < 0.01, Mann–Whitney U test). (B) ATP production was unchanged by cryopreservation (P = 0.38, Mann–Whitney U test). ATP: adenosine triphosphate.
The Positive Staining Area of Insulin Changed After Cryopreservation
Using insulin and DAPI immunofluorescence, the positive insulin staining area was reduced in all cryopreservation groups. Examining the DAPI and merged images, the cells appeared to be randomly reduced in the −80 °C group and CRYOTOP group, while only the central region was visibly reduced in the BICELL group (Fig. 4).
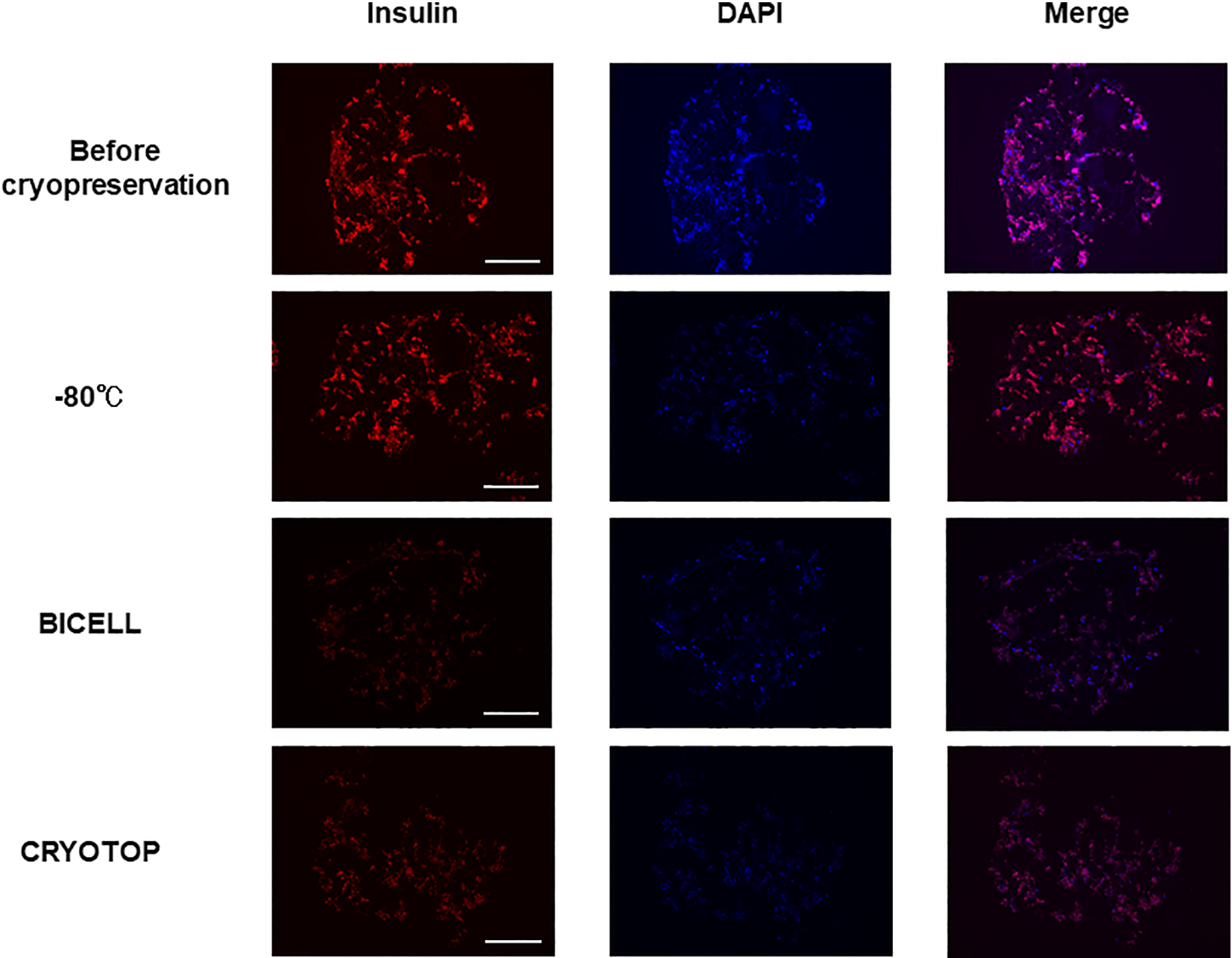
Immunofluorescence staining of insulin in insulin-producing cells. The positive staining area of insulin was reduced in all cryopreservation groups (left line). In DAPI and merged images, the cells appeared to be randomly reduced in the −80 °C group and CRYOTOP group. Meanwhile, the central region was particularly reduced in the BICELL group. Representative images of three independent experiments are shown. Red: insulin; blue: DAPI. Scale bar: 200 µm. DAPI: 4′,6-diamidino-2-phenylindole.
Caspase-3 Antibody Staining of IPCs Remaining After Cryopreservation
IPCs after cryopreservation were strongly stained by cleaved caspase-3 antibody in all three groups, even although naïve IPCs were not stained (Fig. 5).

Caspase-3 immunohistochemistry in insulin-producing cells. Insulin-producing cells remaining after cryopreservation were stained with caspase-3 (arrowheads). R: recombinant peptide pieces. Scale bar: 25 µm.
Decreased Peak Insulin Secretion After Cryopreservation
Finally, the differing range of insulin secretion to glucose stimulation was investigated in IPCs before and after cryopreservation. In all groups, the amount of insulin secretion after freezing was significantly reduced (P < 0.01; Mann–Whitney U test). The average insulin secretion to glucose stimulation was 57.9 pmol/l before cryopreservation, 44.1 pmol/l in the −80 °C group, 39.8 pmol/l in the BICELL group, and 42.8 pmol/l in the CRYOTOP group (Fig. 6).

Glucose-stimulated insulin secretion test. Insulin secretion was decreased significantly by 22 mM glucose stimulation in all groups (P < 0.01, Mann–Whitney U test). Basal insulin secretion (stimulated by 2.2 mM glucose) did not reach significance.
Discussion
The aim of our study was to expand the usefulness of IPC transplantation for patients with T1DM by appropriate cryopreservation. If cryopreservation of IPCs is achievable, it will be possible to wait for the results of required government regulation tests before cell transplantation and transport of IPCs to distant institutes. These possibilities may widely increase their clinical application. However, we found that IPCs after cryopreservation showed a significant decrease in cell quality and cell function. However, several methods for the cryopreservation of islets have already been reported. In one study, islets preserved at 37 °C showed better cell function and survival compared with the −80 °C group on day 1. However, by day 7, they had significantly less function and showed more apoptosis 9 . This suggests that cryopreservation still has disadvantages for β-cell preservation 18 –22 . Moreover, thawed islets are exposed to stresses aside from the thawing process, including the preceding isolation and cryopreservation 23 . Indeed, several studies on post-thawed stress have reported a decline in islet viability after cryopreservation 24 –26 . Furthermore, another study reported that when islets are cooled, cells on the outside experience a greater rate of temperature change than cells on the inside. Because of this differential temperature gradient, ice crystals can form on the inside of cells leading to cellular destruction 27 . As another issue, cryopreservation conditions have not been determined because islets are not composed of homogeneous cells. The rate of freezing and thawing during cryopreservation is important for islet function and morphology. The slower islets are frozen, the more time there is for the liquid inside the cell to reach an equilibrium with the outside of the cell. This prevents destructive ice crystal formation inside the cell 28 –30 . However, we found that the center of cell clusters of generated IPCs did not stain well after Bicell cryopreservation in this study. Central necrosis might occur even when IPCs are frozen slowly. Previously, we have reported that fragile porcine islets can be preserved in a good condition by stepwise lowering of the temperature to 0 °C without freezing 31 . Therefore, IPCs may also be preserved by performing stepwise cooling. Accordingly, we are currently conducting a preservation experiment using this cooling system (data not shown).
In this study, we used three freezing methods: rapid freezing at −80 °C, slow freezing using Bicell, and an embryo cryopreservation method. All methods resulted in significantly reduced viability and quality of IPCs. Furthermore, cryopreserved IPCs were strongly stained by cleaved caspase-3 antibody in all three groups. This staining result suggests that cell death of the IPCs was mainly due to apoptosis. Supporting this finding, a previous study reported reduced recovery rate of islets after storage at −80 °C, which may be associated with apoptosis due to increased hypoxia-inducible factor-1α mRNA expression and protein level of Bax, an apoptotic protein 9 .
Stem cells usually retain a high survival rate even after cryopreservation; however, the results of the present study show that the survival rate decreased when cryopreserved after differentiation into IPCs. Further study and precise comparisons are required to understand the mechanism involved. Although the survival rate of cryopreserved IPCs was markedly reduced, the decline in glucose-stimulated insulin secretion remained approximately 30%. This will be important when considering the transplantation of cryopreserved IPCs. We previously reported that IPCs with poor cell quality did not secrete sufficient amounts of insulin 5,32 and that the transplantation of such IPCs into streptozotocin-induced immunodeficient mice did not improve blood glucose levels 8 . We have already investigated the mechanism of this phenomenon in detail (in press). According to the present study, it could be same results when transplanting IPCs after cryopreservation into mice due to the proven poor cell quality.
To our knowledge, the present study is the first investigation of the cryopreserved IPCs. The ideal cooling method for islets has not yet been completely established, and the mechanism underlying apoptosis has not yet been clarified. Consequently, the cryopreservation of islets is at such a stage that IPCs generated by regenerative medicine require further investigation to overcome their fragility. Because IPCs are homogeneous cells, detailed investigation may lead to identification of the cryopreservation mechanism not only for IPCs but also for islets.
In conclusion, IPCs generated from ADSCs may be too fragile for cryopreservation with accomplished methods because massive cell death occurs that is mainly due to apoptosis. Thus, it is necessary to identify a proper preservation method. Indeed, it is necessary to overcome this fragility urgently, before application of the clinical cryopreservation of IPCs.
Footnotes
Acknowledgments
Ethical Approval
Ethical Approval is not applicable for this article.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partly supported by grants-in-aid for Scientific Research (grant number 19k09045), and by the Japan Insulin-Dependent Diabetes Mellitus (IDDM) network (2019-2021) and Uehara Memorial Foundation (2019). TI and MS have received a research grant from Fujifilm.