
Abstract
NIX/BNIP3L is known as a proapoptotic protein that is also related to mitophagy. Previous reports have shown that NIX could be involved in neuronal apoptosis after intracerebral hemorrhage, but it also plays a protective role in mitophagy in ischemic brain injury. How NIX works in traumatic brain injury (TBI) is unclear. Thus, this study was designed to observe the expression of NIX and perform a preliminary exploration of the possible effects of NIX in a rat TBI model. The results showed that NIX expression decreased after damage, and colocalized with neuronal cells in cortical areas. Moreover, when we induced upregulation of NIX, autophagy was increased, while neuronal apoptosis and brain water content decreased along with neurological deficits. These findings remind us that NIX probably plays a neuroprotective role in TBI through autophagy and apoptosis pathways.
Introduction
Traumatic brain injury (TBI), caused mainly by an external mechanical force, has a specialized and complex pathophysiological process, including not only primary brain injury but also secondary brain injury, which is more closely related to neural functional recovery 1 . The correlated mechanisms such as apoptosis, autophagy, gliosis, inflammation, excitotoxicity and free radical formation are also targets of research for treatment 2,3 .
NIX, also called BNIP3L (Bcl-2/E1B-19K-interacting protein 3-like), belongs to the Bcl-2 family and is considered a proapoptotic protein 4 . It can be activated in many diseases and seems to have different effects in different models 5,6 . NIX-mediated apoptosis can aggravate neuronal damage after intracerebral hemorrhage (ICH) 7 , but contributes to inhibition of tumor growth by accelerating tumor cell apoptosis 8 . The molecular mechanism of NIX-mediated apoptosis is still unknown. In addition to inducing apoptosis by means of its own structure, NIX is also thought to combine with p75NRT (a member of the tumor necrosis factor receptor that is involved in several neuronal populations) 9 , and then induce apoptosis 7 . In further studies, NIX-related mitophagy has been gradually acknowledged 10 , and is another probable pathway for apoptosis 4,11,12 . As previously shown, NIX not only regulates the shift in Parkin and then activates mitophagy by the Parkin-Ubiquitin-p62 pathway 13 , it also induces mitophagy with Atg8-family proteins and serves as an autophagy receptor 14 . In addition, there is also a considerable amount of evidence confirming that NIX-related mitophagy takes part in the pathological process in many diseases 15 –19 . The latest research shows that brain damage was more serious in NIX–/– mice, while more neurons survived after NIX upregulation after cerebral ischemia-reperfusion (I/R) injury 18 . However, there is no report about NIX in TBI models at present. In this study, we aimed to determine how NIX is expressed and the manner in which it works, as these questions are still unclear.
Materials and Methods
Animals and the TBI Model
Male Sprague-Dawley (SD) rats (100 animals, weight 280–320 g) were maintained in a standard animal facility with free access to food and water under 12 h light/dark cycles. All of the animals were obtained from the Animal Center of the Chinese Academy of Sciences (Shanghai, China). The experimental protocols were approved by the Animal Care and Use Committee of Soochow University (Permit Number: 2017-0043) and performed in accordance with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines. A TBI model was produced using the weight-drop injury model, which produces relatively severe injury 20,21 . Each animal was anesthetized with 4% chloral hydrate (400 mg/kg, i.p.). Then, we made a midline incision and conducted a 5 mm craniotomy behind the cranial coronal suture and to the right of the midline. A 40 g weight steel rod was dropped from a height of 25 cm onto another copper weight (4 mm in diameter, 5 mm in height) that was previously placed onto the endocranium of the surgical wound. The impact force of the injury is 1000 g/cm. For the sham group, animals had an operation without the cortical impact. After the surgery, the animals were settled at 37°C on a heating pad until they completely recovered from the anesthetic, and were then returned to their housing cages. The brain tissue surrounding the injured cortex (Fig. 1a) was dissected on ice; some of the tissues were fixed in 4% paraformaldehyde (PFA), and the rest was stored at –80°C until use.

The experimental process. a. Detected areas of brain tissues in the TBI group and the same location in the sham group were showed as the figure, which also indicated in the coronal sections of the damaged brain. b. Experiment I was designed to observe the expression of NIX at different time points after TBI and to find a specific time point for the next experiment. Furthermore, the location of NIX in brain cells was also confirmed at this time point. c. Experiment II was designed to investigate the possible effects of NIX on TBI-induced damage.
Experimental Design
The experiment was designed as follows:
Experiment I
In the first experiment, 48 surviving animals (out of a total of 55) were divided randomly into eight groups according to the different time points after injury. Western blot was used to detect the expression of NIX at 6, 12, 24, 48, and 72 h, and 7 d after TBI in peri-injury cortical tissues. Double-labeling immunofluorescence staining was carried out to observe the cellular localization of NIX in the cerebral cortex with different cellular markers such as a neuronal marker (NeuN), astrocyte marker (glial fibrillary acidic protein (GFAP)), and microglia marker (CD11b) at 24 h after TBI.
Experiment II
Promoting the expression of NIX was the next step. NIX overexpression was induced by intracerebroventricular (ICV) injection at 24 h before TBI modeling. The experimental grouping was as follows: 40 surviving SD rats (out of a total of 45) were divided into the sham group, TBI group, TBI+Over-Ctr group, and TBI+Over-NIX group. Autophagy, nerve apoptosis, and albumin exudation of each group were then measured by Western blot at 24 h after TBI, which was chosen from the result of Experiment I. In addition, other experimental methods such as neurobehavioral scoring, brain water content measurement, TUNEL staining and Fluoro-Jade B (FJB) staining were also performed to observe and compare the differences between groups.
Drug Administration
Animals of group TBI+Over-Ctr and TBI+Over-NIX individually received 20 µl of vehicle or NIX pDNA (500 pmol/10 µL) by ICV injection with a rate of 0.5 µL/min at 24 h post-TBI modeling. Here, the drugs were injected into the right lateral ventricle following a previously described method 22 . Specific pDNA for NIX obtained from Genscript (Nanjing, China) was dissolved in the EntransterTM-in vivo DNA Transfection Reagent (Engreen Biosystem Co, Ltd., Beijing, China) and diluted with 0.9% saline. Vehicle was a mixture of the same concentration of transfection reagent and saline as NIX pDNA.
Western Blot
Rats were administered 4% chloride hydrate and sacrificed. Brain tissues were removed and separated on ice. Peri-injury cortical tissues, which contained the target protein, were immediately frozen at –80°C before use. The sheared tissues were then dissolved by RIPA lysis buffer (Beyotime, Shanghai, China) and phosphatase inhibitors (Abcam, Cambridge, UK) and centrifuged at 13,000 rpm at 4°C for 30 min. The protein concentration of the collected supernatant was tested by a PierceTM BCA Protein Assay Kit (Thermo-Fisher Scientific, Loughborough, UK). The resulting samples were separated by electrophoresis on 10% SDS-polyacrylamide gels (Beyotime, Shanghai, China). After transfer to PVDF membranes (Millipore, Bedford, MA, USA), 5% nonfat milk was used to block the membranes at room temperature for 2 h. The membranes were then incubated with the following primary antibodies overnight at 4°C: rabbit anti-BNIP3 L (1:400, Abcam), rabbit anti-LC3B (1:1000, Abcam), rabbit anti-SQSTM1 (1:1000, Abcam), chicken anti-Albumin (1:1000, Abcam), and rabbit anti-GAPDH (1:10000, Abcam). A corresponding horseradish peroxidase (HRP)-conjugated secondary antibody was used to react with the appropriate primary antibody. Protein bands were developed in an enhanced chemiluminescence system (ECL, Pierce, Waltham, MA, USA) and then analyzed by using ImageJ software.
Double Immunofluorescent Staining
After retrieval of brain tissue, specimens were fixed in 4% PFA for 24 h and dehydrated in 15% and 30% sucrose in turn. Frozen coronal sections of 15 µm thickness embedded in optimal cutting temperature compound (OCT) were prepared for staining. Air-dried slices were washed with 0.3% Triton X-100 three times and blocked with 10% goat serum for 2 h at room temperature for the purpose of reducing nonspecific staining. These slices were incubated overnight at 4°C with the primary antibody anti-BNIP3 L (1:100, Abcam, Cambridge, UK) and the following different markers: mouse anti-NeuN (1:400, Millipore, Bedford, MA, USA), mouse anti-GFAP (1:200, Bio-Rad, Oxford, UK) and mouse anti-CD11b (1:200, Bio-Rad). After incubation with secondary antibodies, 4,6-diamidino-2-phenylindole dihydrochloride (DAPI) was used for staining the cell nuclei. Finally, the sections were observed with a Leica fluorescence microscope (Leica, Wetzlar, Germany) at 200× magnification and photographed by LAS X software.
TUNEL and Fluoro-Jade B Staining
Frozen sections were prepared as described previously and processed by TUNEL Assay Kits (Abcam, Cambridge, UK) according to the manufacturer’s instruction 23 . It was also necessary to incubate the sections with DAPI for 5 min. Cells were observed with a Leica fluorescence microscope (Germany) at 200× magnification. The apoptotic cells were represented by positive cells, and the index of apoptosis was the ratio of (positive cells)/(total cells)×100%.
Injured neurons could be detected by Fluoro-Jade B (FJB) staining. The process of staining was carried out as previously reported 22 . The FJB-positive cells were photographed and measured by LAS X software.
Neurological Score
At 24 h after TBI in experiment II, the degree of nerve injury was evaluated by using the Garcia test 24 (total score of 21), which includes spontaneous activity, body proprioception, response to vibrissae touch, symmetry of limb movement, lateral turning, forelimb outstretching, and climbing ability. Each part of the test was scored from 0 to 3. The lower the score, the more severe the damage. The raters were nonmembers of our group who were double-blinded to the experimental conditions and familiar with the scoring criteria.
Brain Water Content
The wet-dry weight method was adopted to measure the brain water content in experiment II 25 . The fresh brain specimens were divided into two hemispheres and weighed separately (wet weight). Afterward, they were placed into an oven at 100°C for 72 h and then weighed individually (dry weight). The formula to calculate brain water content (%) was [(wet weight–dry weight)/(wet weight)]×100%.
Statistical Analysis
GraphPad software was employed to analyze the data, and all values were expressed as mean±SEM. The comparison between two groups was checked by t-test, and one-way analysis of variance (ANOVA) followed by either a Dunnett or a Tukey post hoc test was used for multiple sample means. p values < 0.05 were considered to indicate statistical significance.
Results
Expression of NIX after TBI
Western blot was performed to determine the expression of NIX in the peripheral cortex (Fig. 1a) at 6 h, 12 h, 24 h, 48 h, 72 h, and 7 d after TBI. As shown in Fig. 2, the NIX protein level was lower in the TBI group than in the sham group. Moreover, there was a noticeable drop in NIX protein level at 24 h in the TBI group. Therefore, we chose this time point for studying the effect of NIX on TBI.

Expression profiles of the NIX protein after TBI. Western blotting was performed to analyze the expression of NIX in the peri-injury cortex at 6, 12, 24, 48, 72 h, and 7 d after TBI. Quantification graphs were produced by using NIX/GAPDH protein levels, and GAPDH was used as an internal standard to confirm equal quantities of protein loading. The results showed that the expression of NIX was decreased after TBI, and, at 24 h, the protein levels began to decrease significantly. Data are shown as the mean±SEM (n = 6, *P < 0.05, **P < 0.01 vs sham).
In double immunofluorescence staining, it was confirmed that NIX colocalized with neurons in the cortex of the brain, and that there were no overlaps with astrocytes and microglial cells (Fig. 3a). Not only the positive-NIX cells (Fig.3c), but also the neurons with positive-NIX in peri-TBI regions were also obviously less than those in sham group at 24 h after TBI (Fig. b,d).
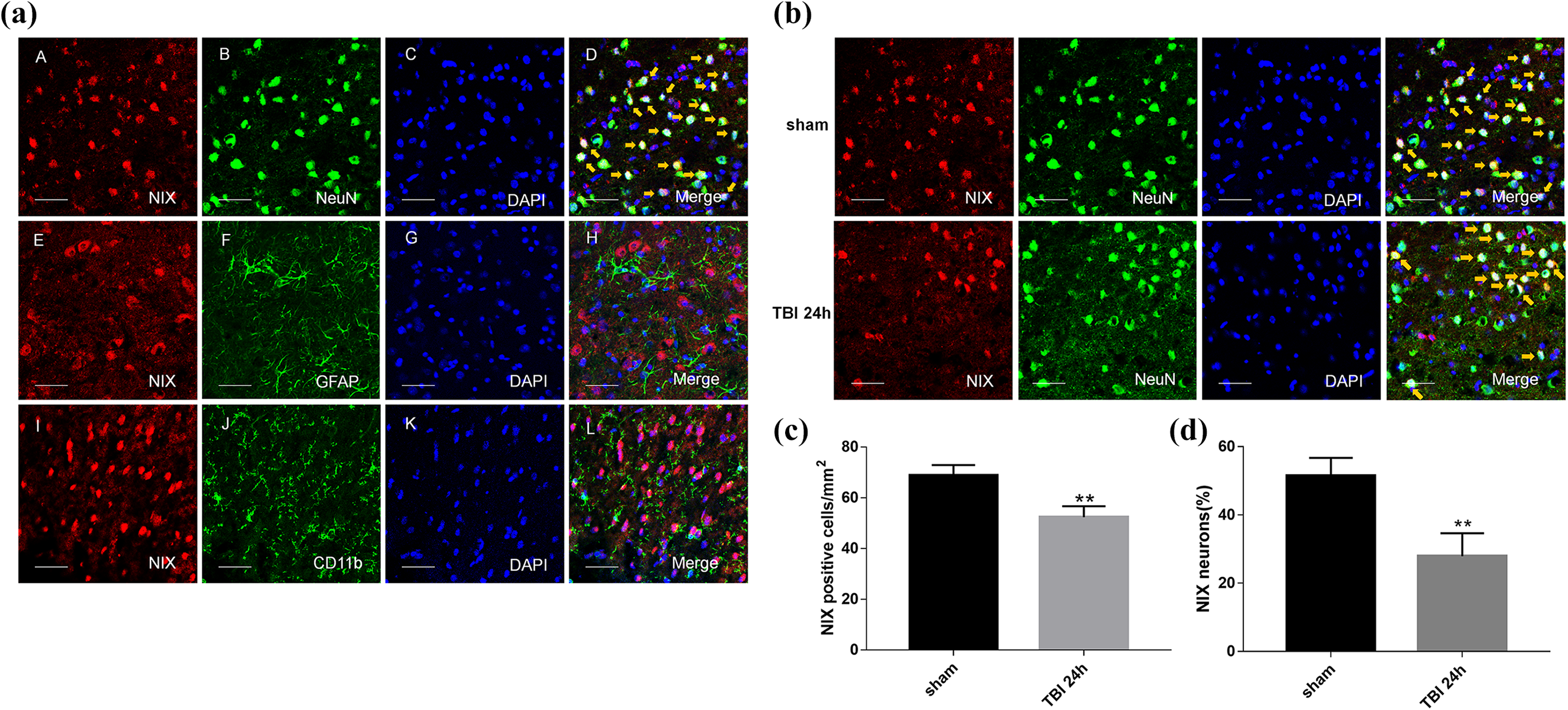
The location of NIX in the cortex of the sham group and changes at 24 h after TBI by double immunofluorescence staining. a. Sections from the sham group were labeled with NIX (red A, E, I), different cell markers (green B, F, J) such as a neuronal marker (NeuN), astrocyte marker (GFAP), and microglial marker (CD11b), as well as DAPI (blue C, G, K) to mark the nucleus. In the merged images (D, H, L), cells shown by the orange arrows could be found in neurons on the whole, but not in astrocytes or microglial cells. b. NIX-positive neurons in the peri-injury cortex at 24 h after TBI were also observed and compared with those in the sham group. c. Quantification of the number of NIX positive cells/mm2 in TBI group and sham group. The results showed that NIX-positive cells were significantly decreased at 24 h after TBI. d. Quantitative analysis was used to verify the differences of NIX-positive neurons between these two groups. Scale bars, 50 µm. Data are shown as the mean±SEM (n = 6, **P < 0.01 vs sham).
Effect of NIX on Brain Water Content and Neurological Function after TBI
To further investigate the effects of NIX in TBI models, NIX pDNA (Over-NIX) was administered via ICV injection at 24 h before TBI modeling to upregulate the expression of the NIX protein. The effect of overexpression was confirmed by Western blot analysis (Fig. 4a).

The changes in brain water content and neurological deficits after overexpression of NIX. a. Western blot and quantitative analysis were used to detect the efficiency of NIX overexpression at 24 h after TBI. b. Then, the brain water content was tested by the wet/dry method as well. The results were compared among each group not only in the ipsilateral hemisphere but also in the contralateral hemisphere.
Meanwhile, administration of NIX significantly reduced brain water contents in the peri-injury brain at 24 h after TBI, and there was no difference between the TBI group and Over-Ctr group. Additionally, the differences in lateral brain water content of each group were not statistically significant (Fig. 4b). As shown in Fig. 4c, although the neurological scores in TBI group and Over-Ctr group were not different from each other, they were apparently lower than those in the sham group. However, the scores could actually be modified in the Over-NIX group compared with those in the Over-Ctr group. It was also apparent that albumin levels tested by Western blot analysis were increased after TBI and that they could be downregulated after NIX overexpression (Fig. 4d).
Effect of NIX on Autophagy and Apoptosis after TBI
Furthermore, autophagy-related proteins such as LC3 and p62 were evaluated at 24 h after TBI. As we had seen in Fig. 5, LC3-II/LC3-I was increased in the TBI and Over-Ctr groups while p62 was decreased. However, compared with the Over-Ctr group, LC3-II/LC3-I was higher, and p62 was lower in the Over-NIX group.

Detection of autophagy changes after overexpression of NIX. a. LC3 protein bands were analyzed by Western blot at 24 h after TBI, and then the ratios of LC3-II with LC3-I were determined by quantification graphs.
Finally, apoptotic cell death was observed by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining (Fig. 6a), and degenerative neurons were detected by Fluoro-Jade B (FJB) staining (Fig. 6b). The results showed that the apoptotic rate and number of degenerative neurons were increased significantly after TBI but were truly reduced when the NIX protein level was upregulated (Fig. 6c).

Detection of apoptosis changes after overexpression of NIX. a. At 24 h after TBI, sections were labeled with TUNEL-activated cells (red) and DAPI (blue), and then, in merged images, cells shown by the orange arrows were counted for statistical purposes. b. FJB-activated cells marked by green were also photographed by LAS X software. Scale bars, 50 µm. c. Quantification graphs of the apoptosis index and FJB-activated cells were drawn separately as described. Data are shown as the mean±SEM (n = 6, **P < 0.01 vs sham, #P < 0.05 vs. Over-Ctr).
Discussion
In this study, we first discovered that the expression of NIX actually decreased at certain times after TBI, with the lowest level appearing at 24–72 h. Furthermore, double immunofluorescence staining showed that NIX basically colocalized with neuronal cells, but not with astrocytes or microglial cells, in the cortex around the injury. These results were not exactly the same as we had expected. In hypoxia, NIX mRNA and protein levels can be upregulated, precisely because of the hypoxia response element (HRE) motif in the NIX promoter region 26,27 . In addition, there is no doubt that hypoxia is the basic pathological mechanism in many brain injury diseases, including cerebral ischemic injury, TBI, and cerebral hemorrhage 28 . Shen et al. observed that the NIX protein level was significantly elevated at 6 h, with the highest level at 48 h after ICH 29 . Similarly, this finding was also observed in ischemia-reperfusion brain injury 18 . Thus, it was only natural to think that the expression of NIX would increase after TBI, but, actually, this was not the case. First, we found that the limitations of time points in our research might be the primary reason for such incidents. The protein levels at 6 h after TBI were still unclear. Second, unlike other ischemic injuries, the mechanical damage caused by TBI can immediately result in the loss of neurons. Lack of a carrier may affect further expression of NIX. Finally, factors other than hypoxia playing a positive role in NIX expression cannot be ruled out. In spite of this, crucially, these problems had no effect on our next experiment and certainly will be investigated in the future.
To further determine the role of NIX after TBI, NIX overexpression plasmids were used to increase the expression of NIX. In view of that significant reduction of NIX at 24 h after TBI in Experiment I, we selected this point to observe overexpression efficiency and conducted the next experiment 24,30,31 . The degree of brain damage and the levels of autophagy and apoptosis were then reassessed. First, it was clearly presented to us that the results of brain water content and neurological scores actually demonstrated the protective effects of NIX. The decreased levels of albumin were also consistent with the above conclusion. Then, the changes in autophagy-related proteins LC3 and p62 were specific to the increased autophagy when the NIX expression level was upregulated. All of these findings reminded us that NIX probably plays a protective role in the autophagy pathway. As we know, autophagy is a selective process of degradation of proteins and organelles by lysosomes 32 . After TBI, neurons will undergo hypoxia, lysis, and swelling 33 . Meanwhile, autophagy is activated and degrades the damaged organelles, proteins, and metabolic waste, which is beneficial for maintaining the stability of the internal environment 32 . There are many studies that agree with the neuroprotective effect of autophagy 34 –36 . As a mitophagy receptor, NIX is involved in mitophagy by many pathways 37 , and its related mitophagy participates in the pathological process of many diseases as well. Whether the increased autophagy was due to NIX-mediated mitophagy was not clear until now and needed further research.
Finally, we were concerned about the decreased level of apoptosis shown by TUNEL and FJB staining in the Over-NIX group compared with the Over-Ctr group. This result was confusing. It is known that NIX-mediated apoptosis was first discovered because of the protein’s own molecular structure, as confirmed by many studies 4 . Previous reports considered that promoting apoptosis might not be the main function of NIX 37 on account of the differences between NIX-mediated apoptosis and typical BH3-only related apoptosis 4,38,39 . Zhang et al. also observed that NIX-mediated apoptosis generally occurred 72 h after hypoxia, later than classical apoptosis 40 . Although NIX can interact with BCL2/BCL-XL or Beclin1 as BH3-only proteins, elevated NIX-related mitophagy possibly disrupts the interaction among them 27,41 . Therefore, it is reasonable to consider that NIX-related mitophagy occurred earlier than NIX-mediated apoptosis or that NIX-related mitophagy might be more powerful than the other form of apoptosis. In addition, this suggestion can explain why autophagy rather than apoptosis was significantly increased when NIX was upregulated. Furthermore, the relationship between autophagy and apoptosis has always been controversial: both can promote programmed cell death; indeed their goal is cell death. The cooperation mode between autophagy and apoptosis is more commonly seen in existing studies. 36,42 But sometimes they are against each other. The time courses of autophagy and apoptosis can be non-uniform 43 –46 . Autophagy can maintain endoplasmic reticulum (ER) function by digesting protein aggregates and misfolded proteins, and then inhibit apoptosis by an ER stress response. Autophagy also provides energy and nutrients to cells by digesting organelles and proteins, thus prolonging cell life. Mitophagy can even prevent apoptosis by reducing mitochondrial outer membrane permeability (MOMP) and mitochondrial pro-apoptotic proteins such as cytochrome C 47 . As Wang et al report, rapamycin (an autophagy inducer) was believed to protect neurons against apoptosis by increasing autophagy and mitophagy 48 . Sevoflurane shows a similar protective mechanism in TBI 49 . In our experiment, we also found that apoptosis decreased when autophagy increased.
Certainly, there were still some limitations in our experiment. In addition to the lack of reports within 6 h after TBI, whether NIX-related mitophagy was actually increased was unknown as well. As time passes, sustained autophagy can aggravate brain damage 50 . How NIX-related mitophagy exerts its effect at later times following TBI also needs to be understood, as does NIX-mediated apoptosis. It is reported that autophagy functions differently with various degrees of damage 51 . Therefore, the effects of autophagy or mitophagy in other forms of TBI-induced damage are likewise ambiguous. In spite of its many limitations, this study still provides potentially useful information and a direction for future research.
In conclusion, NIX plays a neuroprotective role in TBI-induced damage through autophagy and apoptosis pathways, which will probably become a new therapeutic target for the damage after TBI.
Supplemental Material
Supplemental_Figures - Potential Roles of NIX/BNIP3L Pathway in Rat Traumatic Brain Injury
Supplemental_Figures for Potential Roles of NIX/BNIP3L Pathway in Rat Traumatic Brain Injury by Jialing Ma, Haibo Ni, Qin Rui, Huixiang Liu, Feng Jiang, Rong Gao, Yanping Gao, Di Li and Gang Chen in Cell Transplantation
Footnotes
Ethical Approval
Ethical approval to report this case was obtained from Ethic Committee of Soochow University (Permit Number: 2017-0043).
Statement of Human and Animal Rights
All animal studies in this work were approved by the Care and Use Committee of Soochow University and performed in accordance with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.