
Abstract
Introduction
Hepatotoxicity can arise secondary to several medical conditions, including inflammation, sepsis, and therapy. Lipopolysaccharide (LPS) can activate diverse inflammatory pathways and has been implicated in hepatotoxicity. The store-operated calcium entry (SOCE), a key process for maintaining cellular calcium homeostasis, was shown to modulate inflammatory signaling. Reactive oxygen species, which serve a significant role in maintaining cellular function and homeostasis, are often elevated during inflammation, contributing to tissue injury. Therefore, we hypothesized that blocking the SOCE pathway would inhibit LPS-induced hepatotoxicity by suppressing inflammation and oxidative stress.
Methods
To test this, female BALB/c mice were randomly divided into the following experimental groups: control, LPS, LPS + SOCE inhibitor 2-aminoethoxy diphenyl borate (2APB), and 2APB alone. After 24 h of treatment, serum and liver samples were collected from the mice for histopathological, biochemical, and molecular analyses.
Results
Inhibition of SOCE led to a decrease in the elevated liver function enzymes (ALT and AST) and protected the liver parenchymal cells as observed by histopathological assessment. Furthermore, blockade of SOCE significantly suppressed the level of il-1b, il-6, and cox2 genes in the liver tissues of mice treated with LPS. The expression of antioxidant genes (gsta1 and gpx1) was also significantly reduced by LPS treatment, while SOCE inhibition only restored the gpx1 expression. Additionally, treatment with 2APB attenuated LPS-induced oxidative stress in the liver of mice.
Conclusion
Collectively, our work demonstrated the critical involvement of SOCE in regulating inflammation and oxidative stress associated with LPS treatment, thereby reducing hepatotoxicity.
Introduction
The liver serves a vital physiological function for maintaining metabolic activity, preserving energy metabolism, mediating immunity, detoxifying toxins, and synthesizing proteins. Many drugs are extensively metabolized by liver enzymes, making the liver vulnerable to damage by drug overdoses. 1 Furthermore, viral infection, toxins, alcohol, and medication poisoning are major causes of liver diseases and associated death. Annually, about 60,000 cases of liver-related toxicity are caused by drugs in the U.S. 2 Therefore, liver damage caused by medications is a major worldwide health concern that highlights the critical necessity for the development of innovative and efficient therapies to prevent hepatotoxicity.
Inflammation can be initiated as a reaction to certain stimuli, like pathogens, injured cells, or toxins. Inflammatory pathways, which include common inflammatory mediators and regulatory mechanisms, are involved in the pathophysiology of hepatotoxicity. As a result of inflammatory stimuli, multiple signaling pathways are activated, promoting the synthesis of inflammatory mediators, including cyclooxygenase 2 (COX2), interleukin-1 βeta (IL-1β), IL-6, and tumor necrosis factor-alpha (TNF-α). Key intracellular signaling pathways are implicated in the initiation and progression of inflammation, notably signaling pathways of Janus kinase (JAK)/signal transducer and activator of transcription (STAT), the mitogen-activated protein kinase (MAPK), and nuclear factor kappa-B (NF-kB). 3 Dysregulation of these intracellular signaling pathways has been linked to liver diseases.4,5 Upregulation of the COX2 enzyme has been linked with the progression of hepatic fibrosis. 6 Additionally, the regeneration capability of the liver is influenced by TNF-α and IL-6 proinflammatory cytokines.7,8 Toll-like receptor 4 (TLR4), a key component of the TLR family, is essential for initiating and propagating a cascade of inflammatory signaling events. TLR4 is potently activated by LPS, which triggers the production of various proinflammatory mediators, including COX2, IL-1β, IL-6, and TNF-α.9,10 The TLR4 signaling pathway has been linked with the progression of liver diseases.11,12 Genetic deletion of tlr4 mitigated liver injury induced by LPS via suppression of apoptosis and inflammation. 13 Collectively, these data provide strong evidence linking inflammatory signaling to liver diseases and toxicities.
Many inflammatory conditions have been associated with reactive oxygen species (ROS). ROS can result in a variety of consequences, including the activation of NF-kB and the inflammasome. 14 Additionally, Calcium (Ca2+) is released from the endoplasmic reticulum (ER) when intracellular Ca2+ homeostasis is disrupted by ROS imbalance. ROS can stimulate receptors on the ER membrane, such as the ryanodine receptor and the inositol-1,4,5-triphosphate receptor (IP3R), that control intracellular Ca2+ homeostasis, resulting in an efflux of Ca2+ from the ER to activate multiple Ca2+-dependent signaling pathways. The elevated level of cytosolic Ca2+ influences mitochondrial activity, leading to a rise in ROS production from mitochondria. 15
Intracellular Ca2+ concentration is a key element for the proper control of multiple physiological functions. At rest, the Ca2+ is kept at a low concentration in the cytoplasm by storing it in the ER. Furthermore, the movement of Ca2+ through the cell membrane is tightly regulated by multiple Ca2+ channels and transporters. 16 Dysregulation of this finely tuned Ca2+ balance has been linked with some pathophysiological conditions, including liver-related diseases. 17 Depletion of Ca2+ stores in the ER activates a specific Ca2+-related signaling pathway called the SOCE. SOCE is an essential process for maintaining the intracellular balance of Ca2+ by regulating the cellular Ca2+ movement. SOCE is activated when the Ca2+ storage in the ER is depleted. ER Ca2+ sensor proteins, known as STIM1 and STIM2, detect Ca2+ ER store depletion and translocate to activate the cell membrane Ca2+ channels (ORAI1/2/3) to allow the entry of extracellular Ca2+ into the cellular cytoplasm.18,19 A range of normal cellular processes and pathological conditions, including inflammation, is influenced by SOCE. Additionally, SOCE signaling was shown to be critical in modulating TLR4-induced inflammatory signaling in immune and cancer cells.20–22 Several drugs, including SKF-96365, 2APB, and YM-58483, are known to block SOCE channels, although they may also inhibit other signaling pathways.20–22 Collectively, the previous data suggest a link between calcium homeostasis, SOCE, inflammation, oxidative stress and hepatotoxicity. Therefore, the key objective of this study is to evaluate the association between the SOCE pathway and LPS-induced hepatotoxicity through regulating inflammation and oxidative stress.
Materials and methods
Materials
2-APB (SOCE inhibitor), LPS, SYBR Green PCR Master Mix, and cDNA reverse transcription kit were all sourced from Medchemexpress, USA (Catalog No. HY-W009724, HY-D1056, HY-K0522, and HY-K0510A, respectively). TRIzol reagent was obtained from Invitrogen, USA (Catalog No. 15596026). Forward and reverse primers for il-1b, il-6, tnf-α, cox2, sod2, gsta1, and gpx1 genes were designed and synthesized by Integrated DNA Technologies (IDT), Belgium. Colorimetric kits for GPT (ALAT) (REF 12012) and GOT (ASAT) (REF 12011) IFCC mod. liquiUV Humazym Test (HUMAN, Germany), respectively, were used. TBARS assay kit was obtained from Zeptometrix, USA (Catalog No. 0801192). 2APB was dissolved in a solution of 5% dimethyl sulfoxide (DMSO) in normal saline, while LPS was dissolved in normal saline.
Animals
24 female BALB/c mice aged 4 to 6 weeks, weighing between 18 and 22 g, were obtained from the Experimental Surgery and Animal Laboratory, Prince Nayef Research Center, College of Dentistry at King Saud University. Female BALB/c mice are a well-established model to study hepatotoxicity, inflammation, and oxidative stress.23,24 Animals were housed in groups under ideal conditions (25°C temperature and 50-55% humidity) with unrestricted access to diet throughout the study period. No clinical scoring system was utilized to assess animals during the study; however, two humane endpoint criteria, including persistent unresponsiveness or lethargy and signs of severe pain or distress such as difficult breathing and reduced mobility, were set. If mice met these criteria, they would have been sacrificed. However, none of the mice in this study reached these criteria within the study time. Animals were monitored at regular intervals throughout the 24 h of the study. Strict guidelines for conducting animal experiments were followed according to the Ethics of Research on Living Creatures Law issued by the Saudi Council of Ministries, internal regulations of Local committees at the King Saud University, and aligned with the principles of the NIH Guide for the Care and Use of Laboratory Animals (8th edition) and ARRIVE 2.0 guidelines. The ethical approval for this study was formally granted by the Institutional Animal Care and Use Committee under the Scientific Research Ethics Committee at the King Saud University (approval number: KSU-SE-21-83, approval date: 09/12/2021).
Experimental design
The effects of SOCE signaling blockade on LPS-induced hepatotoxicity were assessed by randomly dividing experimental animals, by a technician who was not involved in the analysis of the results, into four groups of six mice each in an unblinded manner as the following: group 1 received 5% DMSO in normal saline without receiving any treatment, group 2 administered a single LPS dose (5 mg/kg),25,26 group 3 received a single 2APB dose (20 mg/kg)27,28 followed 1 hour later by an LPS dose (5 mg/kg), group 4 only received a single 2APB dose (20 mg/kg). LPS and 2APB were administered intraperitoneally (IP) as a single dose. LPS and 2APB doses were selected according to previous in vivo studies conducted in BALB/c mice and other mouse strains.25–29 The 2APB dose (20 mg/kg) has been used earlier in animal studies of other mice strains with good tolerability at this concentration. Therefore, we adopted this dose of 2APB in our study on BALB/c mice, as the physiological similarity between strains supports its use. Furthermore, this dose was shown to be well-tolerated by the mice in our study, with no sign of toxicity. Two mice in the LPS-treated group were lost as a result of LPS-induced toxicity, although they did not show any humane endpoint criteria. These mice were randomly replaced by the same technician to maintain equal numbers of animals across all experimental groups.
The number of animals in each group was not included in all the experiments due to the reagent availability and sample allocation for different experiments. However, independent biological replicates were used in each experiment to ensure statistical validity (N = 6 for the assessment of liver enzymes, N = 3 for histopathology, N = 4 for qRT-PCR, and N = 4 for the TBARS assay), which was supported by previous studies.30–32 Treatment administration, experimental procedures, and data analysis were all conducted in an unblinded manner, which may introduce potential bias. However, the pathologist conducting histopathological examination was blinded to the information, labels, and all other findings. To ensure blinding, slides were coded with identifiers and then handed to the pathologist. Slides were examined randomly and blindly, which minimizes the risk of assessment bias for these results.
Samples collection
Under anesthesia (100 mg/kg of ketamine and 10 mg/kg xylazine, administered IP), the depth of anesthesia was confirmed by monitoring the respiratory pattern. All experimental animals were sacrificed by cervical dislocation after 24 h of treatment, and the death was verified by the absence of reflexes. The blood was harvested from mice through the orbital sinus, and the liver was surgically resected. Serum was obtained from each animal by centrifuging the blood for 10 min at a speed of 3000 rpm, followed by storing serum samples at −80°C. A small portion of each animal’s liver was removed, immersed in 5 mL of 10% neutral buffer formalin (pH 7.4), and then stored at room temperature for histopathology assessment. The remaining liver samples were preserved by placing them in liquid nitrogen for 1 minute, after which they were frozen at −80°C and stored until the time of further molecular analyses.
Assessment of liver function marker levels
The blood serum was used to assess the levels of liver enzymes (AST and ALT). Liver function enzymes were assessed in six biological replicates to provide sufficient statistical power to determine the difference between experimental groups. Detection of the liver enzymes was carried out by a local laboratory in Riyadh, Saudi Arabia, using GPT (ALAT) (REF 12012) and GOT (ASAT) (REF 12011) IFCC mod. liquiUV Humazym Test for ALT and AST (HUMAN, Germany), respectively. 33 The quantification was carried out by measuring absorbance at a wavelength of 340 nm using a Microplate Reader.
Histopathological examination
Liver samples were randomly collected and examined from three biological replicates of each group, which is sufficient to determine histopathological alterations, while conserving samples for further analysis. Samples were sectioned and submitted in histology cassettes for processing. Sectioned specimens were fixed for 48 h in a 10% buffered formalin, followed by paraffin embedding, sectioning (4 μm), and subsequent hematoxylin and eosin (H&E) staining. The histopathological examination was carried out using a Nikon Eclipse Ci-L PLUS Microscope and Nikon Digital Sight Fi3 camera with NIS-Elements Imaging Software, and images were captured at 40X objective (400x magnification). 50 fields were examined, and representative fields were chosen as images. The pathologist was blinded to the information, labels, and all other results; slides were examined blindly and randomly.
Histopathological evaluation was performed using the Batts-Ludwig grading and staging system, which is the most commonly used method for assessing liver injury. 34 The system utilizes 0-4 grades to determine the amount of portal/periportal and lobular activity, 0 indicating none or confined to portal tracts, 1 indicating minimal, patchy interface hepatitis, 2 indicating mild interface hepatitis involving some or all portal tracts, 3 indicating moderate interface hepatitis involving all portal tracts, and 4 indicating severe interface hepatitis/bridging necrosis. Grades for Lobular activity are denoted as 0 for none, 1 for minimal with rare spotty necrosis, 2 for mild with scattered necrotic hepatocytes, 3 for moderate with confluent necrosis and clusters of dead hepatocytes, and 4 for severe with bridging necrosis. The amount of fibrosis is staged from 0 to 4 as follows: 0 referring to no increased fibrous tissue, 1 referring to fibrous portal expansion, 2 referring to periportal fibrosis with periportal septa, 3 referring to bridging fibrosis with portal/portal fibrous septa and distorted architecture, and 4 referring to probable or definite cirrhosis with nodule formation.
Quantitative real-time polymerase chain reaction (qRT-PCR)
List of genes utilized in the study with their gene bank accession numbers and nucleotide sequences of primers.

Thiobarbituric acid reactive substances (TBARS) assay
The TBARS assay, a commonly utilized method to quantify lipid peroxidation, was done according to the manufacturer’s guidelines (Zeptometrix, USA) on four biological replicates per group to maintain sufficient statistical power and optimal use of materials, reagents, and samples for further analysis. 2.5 mL of the thiobarbituric acid reagent was added to the homogenized liver tissues, and the samples were then heated to 95°C for an hour. After that, the samples were cooled down to room temperature, followed by centrifugation for 15 min at 1200×g. A microplate reader was utilized to measure the absorbance.
Statistical analysis
Each experiment was conducted on independent biological replicates. GraphPad Prism version 9 (GraphPad Software, USA) was acquired to perform statistics and graphing. Data were displayed as mean ± SEM. To calculate the statistical difference between groups, a one-way analysis of variance (ANOVA) was performed with the Tukey post hoc test for multiple comparisons. 36 The difference between experimental groups was deemed statistically significant at a threshold of p < 0.05.
Results
SOCE inhibitor 2APB attenuated the LPS-induced elevation of the liver enzymes in mice
An elevation in the levels of serum AST and ALT can be a sign of liver toxicity. Therefore, the levels of these enzymes were evaluated to examine the influence of SOCE blockade on LPS-associated hepatotoxicity. Treatment with LPS significantly increased AST and ALT levels compared to the control group (Figures 1(A) and (B)). These elevations in hepatic enzymes were reduced significantly by combining 2APB with LPS. 2APB alone did not significantly affect the level of these enzymes relative to the control. The influence of 2APB on LPS-induced elevation of mice liver enzymes. Biochemical analysis of serum ALT and AST using serum samples. (A) Serum AST levels. (B) Serum ALT levels. Asterisks (*) and hash (#) symbols denote statistically significant differences from control and LPS, respectively. Data were presented as mean ± SEM, and statistical comparison between groups was conducted using a one-way ANOVA followed by the Tukey post hoc test. Significance levels: ** and *** refer to p < 0.01 and p < 0.001 versus the control group, respectively, and #, ##, ###, and #### refer to p < 0.05, p < 0.01, p < 0.001, and p < 0.0001 versus the LPS group, respectively. N = 6/group. Abbreviations: AST: aspartate aminotransferase; ALT: alanine transaminase; LPS: lipopolysaccharide; 2APB: 2-aminoethoxy diphenyl borate.
SOCE inhibitor 2APB attenuated LPS-induced hepatotoxicity
The liver architecture of mice was examined to determine the impact of SOCE inhibition on LPS-induced hepatotoxicity. Histopathological evaluation was performed using the Batts-Ludwig grading and staging system.
34
Liver sections from control mice displayed normal histological architecture, characterized by consistent hepatocytes and the absence of chronic inflammation, acute inflammation, or portal edema. Additionally, portal/periportal and lobular activity are grade 0, and fibrosis is stage 0 in the control group (Figure 2(A)). In contrast, liver sections from LPS-treated mice showed grade 2 portal/periportal activity, grade 3 lobular activity, and stage 0 fibrosis. Furthermore, the liver of LPS-treated mice exhibited necrosis, vacuolated hepatocytes at the periphery, and acute severe inflammation with interface hepatitis (Figure 2(B)). Mice co-treated with LPS and 2APB showed minimal inflammation, with no evidence of portal edema or acute inflammatory infiltrates, with grade 1 for portal/periportal activity, grade 0 for lobular activity, and stage 0 for fibrosis (Figure 2(C)). Liver sections from mice treated with 2APB alone showed no signs of chronic inflammation, acute inflammation, or portal edema, with grade 0 for portal/periportal and lobular activity stage 0 for fibrosis (Figure 2(D)). Representative photographs of histopathological changes in mice liver sections for different experimental groups. (A) Liver section of control mice. (B) Liver section of LPS-treated mice (5 mg/kg). (C) Liver section of LPS- and 2APB-treated mice. (D) Liver section of 2APB-treated mice (20 mg/kg). Arrow: branch of bile duct; Dot: portal venule; square: hepatocytes; arrowhead: arteriole; circle in B: acute severe suppurative inflammation with interface hepatitis involving some portal tracts (40%); circle in C: monocyte infiltration; and star: confluent necrosis with severe acute suppurative inflammation (Magnification 400x). H&E, scale bar = 50 µm. N = 3/group.
The SOCE inhibitor 2APB blocked LPS-induced expression of proinflammatory mediators in the mouse liver
il-1b, il-6, tnf-α, and cox2 genes were evaluated across various experimental groups. LPS significantly elevated the expression of il-1b and il-6 genes compared to the control (Figure 3(A)–(B)) with no effect on tnf-α (Figure 3(C)). However, the concurrent administration of 2APB and LPS led to a reduction in the expression of these genes. Notably, 2APB alone did not impact il-1b, il-6, and tnf-α gene expression relative to the control group. Additionally, the levels of hepatic cox2 gene were evaluated in various experimental groups. The expression of the cox2 gene was significantly increased upon treatment with LPS compared to the control group (Figure 3(D)). Cotreatment of LPS and 2APB significantly reduced the cox2 gene compared to LPS alone. 2APB alone had no influence on the expression of the cox2 gene. The influence of 2APB on LPS-induced the level of inflammatory genes in the mice liver. The level of il-1b, il-6, tnf-α, and cox2 mRNA was evaluated using qRT-PCR. (A-D) Gene expression of il-1b, il-6, tnf-α, and cox2, respectively. Asterisks (*) and hash (#) symbols denote statistically significant differences from control and LPS, respectively. Data were presented as mean ± SEM, and statistical comparison between groups was conducted using a one-way ANOVA followed by the Tukey post hoc test. Significance levels: ** and *** refer to p < 0.01 and p < 0.001 versus the control group, and ## and ### refer to p < 0.01 and p < 0.001 versus the LPS group, respectively. N = 4/group. Abbreviations: il-1b: interleukin-1 βeta; il-6: interleukin-6; tnf-α: tumor necrosis factor-alpha; cox2: cyclooxygenase 2; LPS: lipopolysaccharide; 2APB: 2-aminoethoxy diphenyl borate.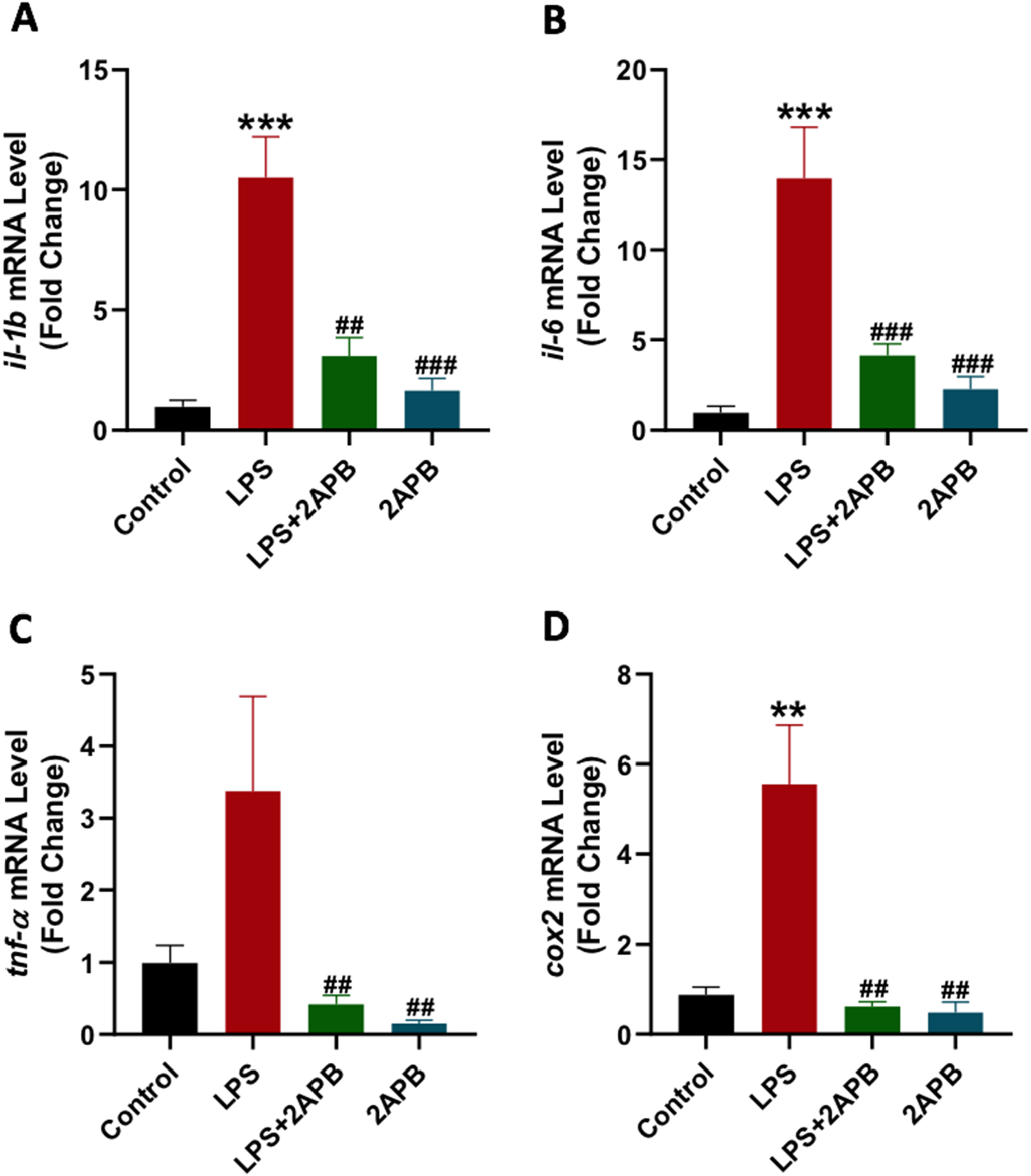
The SOCE inhibitor 2APB restored the antioxidant activity in the mice’s liver following LPS treatment
To assess the influence of SOCE inhibition on oxidative stress, antioxidant gpx1, gsta1, and sod2 genes were measured. LPS administration significantly decreased the level of gpx1 (Figure 4(A)) and gsta1 (Figure 4(B)) genes compared to the control. The reduction in gene expression of gpx1 was significantly restored by combining 2APB with LPS, but not for gsta1. The LPS impact on the sod2 gene showed a similar pattern of reduction, but this effect was not statistically significant (Figure 4(C)). 2APB alone had no effect on the gpx1 gene compared to the control; although, 2APB significantly decreased the gsta1 and sod2 gene expression relative to the control. The influence of LPS and 2APB on antioxidant genes in the mice liver. The mRNA expression of gpx1, gsta1, and sod2 was quantified using qRT-PCR. (A-C) The expression of gpx1, gsta1, and sod2 genes, respectively. Asterisks (*) and hash (#) symbols denote statistically significant differences from control and LPS, respectively. Data were presented as mean ± SEM, and statistical comparison between groups was conducted using a one-way ANOVA followed by the Tukey post hoc test. Significance levels: *, **, and *** refer to p < 0.05, p < 0.01, and p < 0.001 versus the control group, respectively, and # and ## refer to p < 0.05 and p < 0.01 versus the LPS group, respectively. N = 4/group. Abbreviations: gpx1: glutathione peroxidase; gsta1: glutathione s-transferase alpha 1; sod2: superoxide dismutase 2; LPS: lipopolysaccharide; 2APB: 2-aminoethoxy diphenyl borate.
The SOCE inhibitor 2APB blocked LPS-induced oxidative stress in the mice liver
The TBARS level, which quantifies lipid peroxidation, was measured on multiple experimental conditions. LPS resulted in a statistically significant elevation in the TBARS levels in the hepatic tissues compared to the control (Figure 5). Concurrent treatment with 2APB mitigated the LPS-induced increase in TBARS levels. 2APB alone did not affect the level of TBARS relative to the control group. The Effect of 2APB on LPS-induced oxidative stress in the mice liver. Biochemical analysis of TBARS in the liver. Asterisks (*) and hash (#) symbols denote statistically significant differences from control and LPS, respectively. Data were presented as mean ± SEM, and statistical comparison between groups was conducted using a one-way ANOVA followed by the Tukey post hoc test. Significance level: **** refers to p < 0.0001 versus the control group and #### refers to p < 0.0001 versus the LPS group. N = 4/group. Abbreviations: MDA: malondialdehyde; LPS: lipopolysaccharide; 2APB: 2-aminoethoxy diphenyl borate.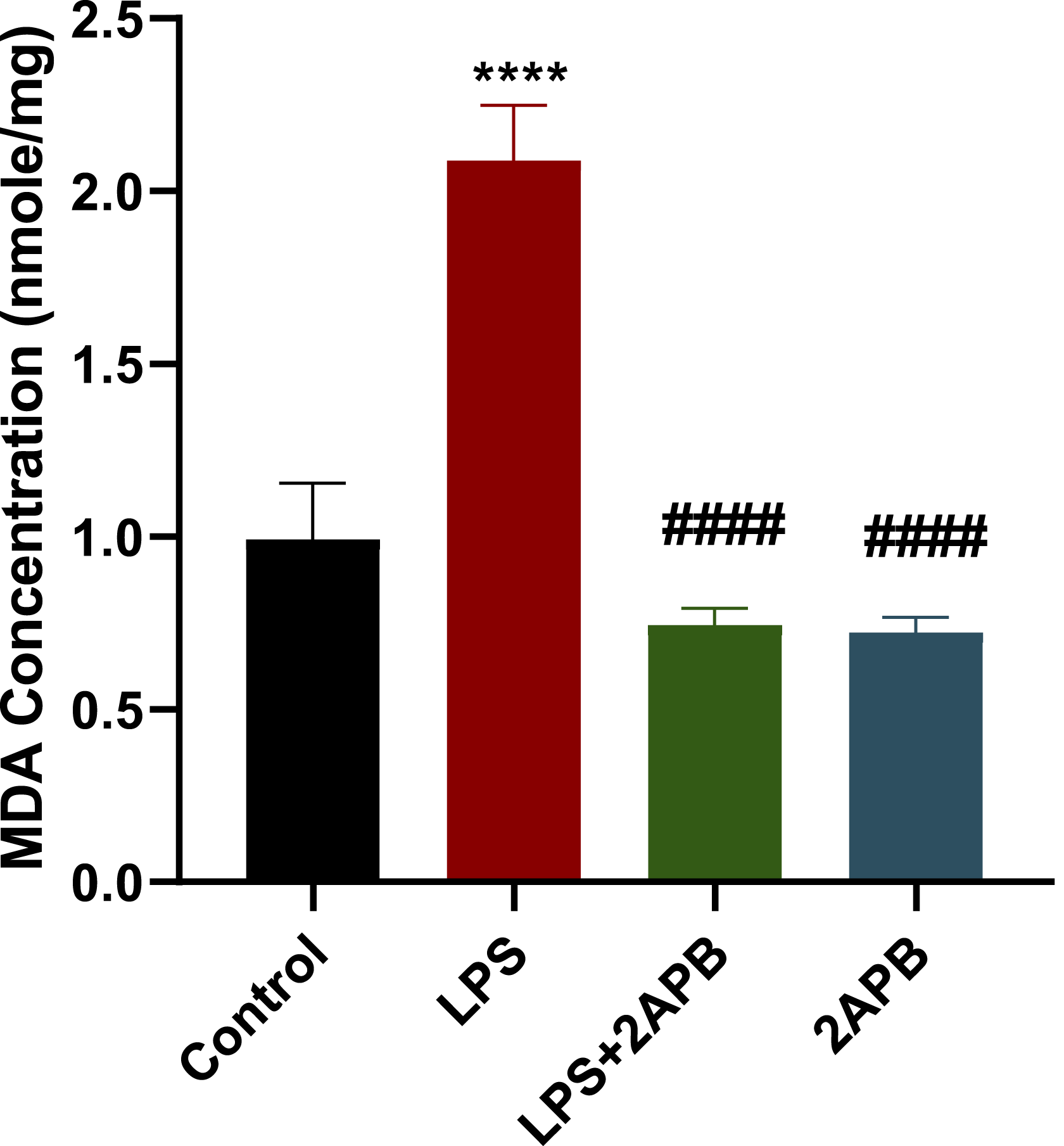
Discussion
Liver-related morbidity and mortality are one of the major medical concerns, often caused by viral infection, toxins, excessive alcohol intake, and drug poisoning. 2 Such challenges emphasize the urgency of developing innovative and effective treatments to protect against drug-induced hepatotoxicity. Inflammation is among the main mechanisms responsible for the development of hepatotoxicity during toxic insults.4–6,8 SOCE has been implicated in inflammation, and blocking this pathway was shown to suppress inflammation across various experimental disease models.19,21,22 Therefore, the current study was designed to assess the effect of the SOCE inhibition on LPS-induced hepatotoxicity.
This study aimed to assess the prophylactic potential of SOCE inhibition against LPS-induced inflammatory and oxidative stress responses, thereby reducing hepatotoxicity. Therefore, 2APB was administered 1 hour before the treatment with LPS. Prophylactic strategy has been commonly used in clinical practice to prevent drug-induced toxicity, for example, the use of mesna and dexrazoxane to prevent cyclophosphamide-induced hemorrhagic cystitis and doxorubicin-induced cardiotoxicity, respectively.37,38 Nevertheless, posttreatment efficacy of 2APB on LPS-induced hepatotoxicity should also be evaluated, which would offer an additional clinical translational significance.
This study intended to evaluate whether the inhibition of SOCE is sufficient to confer protection against LPS-induced hepatotoxicity. Our findings showed that the levels of ALT and AST were elevated after LPS treatment and attenuated by SOCE inhibition. Moreover, histopathological analysis revealed that SOCE inhibition markedly reduced hepatic inflammation induced by LPS, in contrast to necrosis and acute severe inflammation observed in the LPS-treated mice. In line with our findings, previous studies showed that SOCE inhibition attenuated LPS-induced lung and kidney injuries.39,40 Collectively, these findings provide evidence that blockade of SOCE signaling alleviates LPS-induced liver damage and highlights its possible role as a target for mitigating liver injury.
The current study showed that LPS induced an upregulation in the production of inflammatory markers, including il-1b, il-6, and cox2, in mouse liver tissue. These findings are similar to prior studies demonstrating that LPS enhanced the production of inflammatory cytokines across various experimental models.22,41 Notably, our results revealed that the SOCE inhibition effectively blocked the LPS-induced hepatic inflammation. The SOCE inhibitor BTP2 has previously been shown to suppress LPS-induced inflammatory signaling in immune and breast cancer cells.20,22 Furthermore, blockade of SOCE has been reported to enhance the sensitivity of breast cancer cells to chemotherapy via suppressing the associated inflammatory response. 21 In line with our findings, Wasnik et al. showed that SOCE inhibition mitigated LPS-induced mouse renal inflammation by decreasing the level of nfat-1, nf-kb, tnf-α, il-1b, il-6, il-17, and il-18 genes. 39 Similarly, in LPS-induced acute lung injury animal model, SOCE inhibition markedly suppressed the expression of inflammatory genes, nfat-1, nf-kb, tnf-α, ifn-γ, il-1b, il-6, and il-17, caused by LPS treatment. 40 While these studies focused on LPS-induced lung and renal toxicities, the current study focuses on the impact of the pharmacological inhibition of SOCE in LPS-induced hepatotoxicity. Compared to the lung and kidney, the liver is a particularly more vulnerable organ to drug-induced toxicities and associated inflammation and oxidative stress due to its function as a primary organ in drug metabolism and detoxification. In the context of hepatic ischemia-reperfusion injury animal model, tnf-α, il-6, il-1b, inos, cox2, and nfat-1 were upregulated. However, genetic knockdown of stim1 significantly decreased these inflammatory cytokines compared to wild-type mice. 42 Furthermore, STIM1 and ORAI1 exaggerated LPS-induced inflammatory response in bovine hepatocytes, while BTP2 reversed this effect. 41 Although these studies were conducted in the liver, the used models were either in vitro or hepatic ischemia-reperfusion injury, whereas our work utilized an LPS-induced hepatotoxicity animal model. Thus, our study provides further evidence that SOCE can be a valuable target for therapeutic purposes by suppressing inflammatory signaling and protecting against liver injury.
The current study focused on the pathophysiological responses of LPS and 2APB treatments, and the molecular mechanisms mediating these effects have not been examined yet. One possible mechanism that might mediate the hepatoprotective effect of 2APB is via the calcineurin/NFAT signaling pathway. In this pathway, LPS activates TLR4 in liver cells, such as Kupffer cells and hepatocytes, and stimulates SOCE. 43 The calcium entering the cells via the SOCE process activates calcineurin, a phosphatase enzyme that dephosphorylates NFAT. 44 The dephosphorylation of NFAT activates and triggers its translocation from the cytoplasm to the nucleus. 44 In the nucleus, NFAT enhances the production of many genes that mediate inflammation, 45 oxidative stress, 46 and cell death, ultimately contributing to liver injury.47,48 2APB, via blocking SOCE, can therefore inhibit the LPS-induced activation of the calcineurin/NFAT signaling pathway and the subsequent hepatotoxicity. Future studies can test this theory by examining the calcium levels, calcineurin phosphatase activity, NFAT phosphorylation levels, and NFAT translocation after LPS and 2APB treatments to evaluate whether the protective effect of 2APB is mediated via this pathway. Another potential mechanism that can mediate the protective effect of 2APB against LPS-induced hepatotoxicity is by inhibiting the NF-κB pathway. LPS, via TLR4 activation, initiates the NF-κB signaling by stimulating the IKK complex, which phosphorylates and degrades IκBα, thereby allowing p65/p50 subunits to translocate into the nucleus to stimulate transcription of several inflammatory genes. 49 This pathway has been heavily implicated in hepatotoxicity.12,13 SOCE activates this pathway by stimulating protein kinase C (PKC), which subsequently activates the IKK complex, thereby initiating the NF-κB pathway. 50 Therefore, 2APB can suppress LPS-induced NF-κB signaling and the production of inflammatory genes, thereby protecting against LPS-induced liver injury. IκBα phosphorylation/degradation and p65 nuclear localization can be examined to determine whether 2APB indeed influences this pathway during LPS treatment.
The process of SOCE requires both ORAI channels and calcium-sensor STIM proteins. However, differences exist between different isoforms of ORAIs and STIMs. ORAI1 and STIM1 were shown to play major roles in SOCE in hepatocytes and mediate LPS-induced SOCE and inflammatory signaling in these cells. 43 Interestingly, 2APB strongly inhibits ORAI1 but slightly suppresses ORAI2 activity, suggesting that observed 2APB’s protective effects are mediated via targeting ORAI1 compared to other isoforms. 51 However, further sophisticated analysis is needed to gauge the precise roles of ORAI1, ORAI2, ORAI3, STIM1, and STIM2 in mediating the 2APB effects in the LPS-induced hepatotoxicity model. This can be achieved by conducting gene editing experiments to create ORAI and STIM knockout models to assist in examining the role of each protein.
In addition to inflammation, oxidative stress and ROS production are heavily linked with liver diseases.52,53 Recent studies have linked SOCE with oxidative stress and antioxidant defense mechanisms. 54 Therefore, another aim of the current study was to explore the role of SOCE in modulating LPS-induced oxidative stress in the liver. Our findings revealed that the hepatic expression of key antioxidant genes, including gpx1 and gsta1, was reduced by treatment with LPS, while increased TBARS level was observed compared to the control mice. Interestingly, SOCE inhibition restored the gpx1 expression and reduced the oxidative stress level caused by LPS, but had no significant impact on sod2 and gsta1. This might be explained by the differences in the transcription factors driving the expression of each of these genes. For example, GSTA1 transcription is predominantly activated by NRF2, 55 while SOD2 expression is controlled by NF-κB. 56 Meanwhile, ATF4 activates the transcription of GPX1. 57 Interestingly, SOCE has been shown to stimulate NRF2, 58 NF-κB, 43 while inhibiting ATF4. 59 The fact that SOCE can differentially regulate these transcription factors may explain why the SOCE inhibitor used in this study, 2APB, amplified the expression of gpx1 but reduced the basal level of sod2 and gsta1 genes. Another possibility is that 2APB targeted another pathway that is unrelated to SOCE but is important for the gene production of sod2 and gsta1. Consistent with our findings, pharmacological and genetic inhibition of SOCE attenuated oxidative stress-induced apoptosis in normal human melanocytes. 60 In calf isolated hepatocytes, both BTP2 and siRNA-mediated ORAI1 silencing enhanced GSH and SOD activity, while decreasing the concentration of malondialdehyde, hydrogen peroxide, and ROS. 61 Similarly, in hepatic ischemia-reperfusion injury murine model, the observed elevation in oxidative stress in liver tissue was reversed by stim1 genetic knockdown, as shown by the increase in SOD activity and the decrease in the ROS and malondialdehyde levels. 42 The crosstalk between SOCE and calcium signaling in the mitochondria is well-established. 62 Calcium entering the cells via SOCE can be transported into the mitochondria, which increases the ROS production from mitochondria.54,63 The elevation of ROS levels in the mitochondria is linked to NF-κB and NLRP3 inflammasome activation, and elevations in ALT and AST.64–66 Therefore, some of the effects of 2APB might be attributed to its ability to block SOCE-induced ROS production from the mitochondria and the subsequent NF-κB and NLRP3 inflammasome activation, inflammatory cytokine production, as well as AST and ALT elevations. These data indicate that SOCE may play a vital role in mitigating hepatotoxicity induced by oxidative stress and inflammation with a possible involvement of the mitochondria.
While the current study has not directly assessed the impact of SOCE inhibition using 2APB on calcium electrophysiology, 2APB has been previously confirmed to effectively reduce Ca2+ influx in various experimental models. For example, Nicoud et al. (2007) reported that 2APB blocked Ca2+ cellular accumulation and reduced Ca2+ uptake in liver mitochondria of HepG2 cells in a dose-dependent manner. 67 2APB, at a high concentration, has also been confirmed to efficiently block SOCE in breast cancer cells. 68 Similarly, 2APB has been shown to block SOCE in HEK293 cells by preventing STIM1 localization to its functional sites near the cell membrane. 69 We acknowledge the lack of 2APB selectivity on SOCE, as it can modulate other targets, such as IP3 receptors and certain transient receptor potential ion channels. Therefore, the current study cannot exclude the contribution of other pathways to the observed hepatoprotective effect, and this issue should be addressed in future research. Nonetheless, the selective SOCE inhibitor, BTP2, has been shown to protect against LPS-induced kidney and lung toxicity39,40 via suppressing the inflammatory response, supporting our conclusion that inhibiting SOCE is highly likely responsible for the observed hepatoprotective effect. Therefore, the utilization of structurally unrelated SOCE inhibitors or ORAIs and STIMs liver-targeted knockdown/conditional KO can be useful techniques in future studies to clearly delineate which of the 2APB effects observed here are attributed specifically to SOCE inhibition.
Data from our study, combined with previous studies, indicate that SOCE could be a potential therapeutic target in the management of drug-induced hepatotoxicity, especially drugs that induce liver toxicity via inflammatory signaling, oxidative stress, and calcium overload. Several members of antibiotics, antidepressants, chemotherapeutics, and other drug classes trigger inflammation, oxidative stress, and hepatotoxicity, making them a suitable target for SOCE inhibitors. 70 However, a more suitable candidate that can be an alternative to 2APB may be required to enhance specificity and safety. For example, the SOCE inhibitor Auxora has shown promising efficacy and safety in clinical trials for patients suffering from COVID-19 pneumonia. 71 This can be translated to study the safety and efficacy of this SOCE inhibitor in hepatotoxicity, especially against drug-induced liver injury resulting from inflammation and calcium overload.
Although the current study offers valuable insights into the protective role of SOCE targeting against hepatotoxicity through modulation of inflammatory and oxidative stress responses, several limitations remain to be addressed. LPS mainly exerts its effect via TLR4; however, the current study has not directly assessed TLR4 signaling, which can be a potential avenue to explore in future studies. Mainly, further research is needed to confirm that the effects observed with 2APB treatment are due specifically to its action on SOCE rather than other targets. Further, future studies are needed to elucidate the underlying molecular mechanisms responsible for the observed effect of targeting SOCE. Additionally, since the current study utilized an acute inflammation model, further investigations are recommended to evaluate the effect of SOCE inhibition in models of chronic inflammation. The duration of LPS treatment used in this study, which was 24 h, has some advantages and limitations. For example, serum levels of AST and ALT were shown to peak after 24 h of LPS treatment. 72 Similarly, histopathological signs of hepatotoxicity, such as necrosis and inflammation, as well as oxidative stress, can be observed following 24 h of LPS treatment. 72 As for the inflammatory cytokine production, the levels of many of these cytokines typically peak at earlier timepoints; however, many cytokines, including Il-1β and Il-6, remain significantly elevated and detectable 24 h after LPS treatment. 73 Hence, many studies, including ours, used this time point to study LPS-induced hepatotoxicity.74,75 Nevertheless, this should be taken into account when interpreting the results of the study, and further future studies that include different durations of treatments may provide additional information on the effects of LPS and 2APB, especially regarding inflammatory gene production.
Conclusion
In conclusion, this study suggests that blockade of the SOCE pathway effectively mitigates LPS-induced hepatotoxicity by alleviating inflammation and oxidative stress, thereby restoring hepatic enzyme activity and preserving hepatic parenchymal structures (Figure 6). These data emphasize the possible role of SOCE as a valuable therapeutic target for preventing and managing hepatotoxicity and the associated inflammation and oxidative stress. Schematic summary of the study experimental design and main findings. Female BALB/c mice were divided into four groups: control, LPS, LPS+2APB, and 2APB alone. Following 24 h of treatment, the blood and liver were isolated for further analyses. The blood samples were analyzed to assess the levels of ALT and AST after treatment, whereas liver samples were utilized to conduct TBARS assay, gene expression analysis using qRT-PCR, and histopathological assessment. LPS significantly enhanced the levels of AST, ALT, MDA concentration, and inflammatory genes. The LPS-induced elevation of these markers was inhibited by 2APB. LPS treatment also suppressed the gene expression of the antioxidant gsta1; however, 2APB blocked the LPS-induced suppression of gsta1. Finally, histopathological analysis showed that LPS caused damage to liver architecture, while 2APB protected the liver from the LPS-induced damage. This figure was generated with permission using BioRender. Abbreviations: AST: aspartate aminotransferase; ALT: alanine transaminase; il-1b: interleukin-1 βeta; il-6: interleukin-6; cox2: cyclooxygenase 2; gsta1: glutathione s-transferase alpha 1; MDA: malondialdehyde; LPS: lipopolysaccharide; 2APB: 2-aminoethoxy diphenyl borate.
Footnotes
Acknowledgments
The authors extend their appreciation to the Ongoing Research Funding Program (ORF-2025-834), King Saud University, Riyadh, Saudi Arabia for funding this research work.
Ethical approval
Strict guidelines for conducting experiments and procedures on animals were followed in accordance with Ethics of Research on Living Creatures Law issued by Saudi Council of Ministries, internal regulations of Local committees at the King Saud University, and aligned with the principles of the NIH Guide for the Care and Use of Laboratory Animals (8th edition) and ARRIVE 2.0 guidelines. The ethical approval of the animal study was formally granted by the Institutional Animal Care and Use Committee under the Scientific Research Ethics Committee at the King Saud University (reference number: KSU-SE-21-83, approval date: 09/12/2021).
Author contributions
Conceptualization, Abdullah S. Alhamed and Mohammed Alqinyah; methodology, Abdullah S. Alhamed, Mohammed Alqinyah, Khalid Alhazzani, Mohammad M. Algahtani, Hussain N. Alhamami, Ahmed Z. Alanazi, and Faris Almutairi; software, Abdullah S. Alhamed and Mohammed Alqinyah; validation, Abdullah S. Alhamed, Mohammed Alqinyah, Khalid Alhazzani, Mohammad M. Algahtani, Hussain N. Alhamami, Ahmed Z. Alanazi, and Faris Almutairi; formal analysis, Abdullah S. Alhamed and Mohammed Alqinyah; investigation, Yasseen A. Alassmrry, Mohamed Mohany, Mohammad M. Algahtani, and Khaldoon Aljerian; resources, Abdullah S. Alhamed, Mohammed Alqinyah, Khalid Alhazzani, Khaldoon Aljerian, Ahmed Z. Alanazi, and Faris Almutairi; data curation, Abdullah S. Alhamed, Mohammed Alqinyah, and Yasseen A. Alassmrry; writing—original draft preparation, Abdullah S. Alhamed and Mohammed Alqinyah; writing—review and editing, Abdullah S. Alhamed, Mohammed Alqinyah, Khalid Alhazzani, Mohammad M. Algahtani, Hussain N. Alhamami, Ahmed Z. Alanazi, and Faris Almutairi; visualization, Abdullah S. Alhamed and Mohammed Alqinyah; supervision, Abdullah S. Alhamed and Mohammed Alqinyah; project administration, Abdullah S. Alhamed and Mohammed Alqinyah; funding acquisition, Abdullah S. Alhamed. All authors have read and agreed to the published this version of the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the Ongoing Research Funding Program (ORF-2025-834), King Saud University, Riyadh, Saudi Arabia.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
All data of the current study have been included in this published article and are available upon request from the corresponding authors.