
Abstract
Introduction
Alcohol (ethanol or ethyl alcohol), a commonly consumed recreational beverage in most parts of the world, is a hepato-toxicant and a dependence-producing psychotropic substance. 1 Almost 50% population of the world consumes alcohol, and alcoholism increases a grave threat to public health as it leads to numerous physiological dysfunctions and pathological disorders that include hepatic, gastrointestinal, nervous, and cardiovascular injuries. Globally, alcohol consumption is as a major cause of advanced diseases, disability, and death which may result from liver damage. 2 The liver is an essential organ that performs various key metabolic activities to detoxify and depose several xenobiotics.3,4 Ethanol is a xenobiotic metabolized primarily in the liver and therefore is the main target organ for damage done by ethanol. Exorbitant or chronic use of ethanol is responsible for developing a wide range of alcohol-induced liver diseases characterized by fatty liver, hyperlipidemia, inflammation of the liver, fibrosis, hepatic cirrhosis, malignant hepatoma, and liver failure.5,6 Ethanol in the liver is first processed to acetaldehyde through a complex process of catabolic pathway and then into acetate involving the enzymes such as acetaldehyde dehydrogenase (ALDH) and alcohol dehydrogenase (ADH). 7 In hepatic cells, acetaldehyde, and acetate can form additional products with essential macro-molecules, for instance, proteins and DNA, thereby resulting in defective structure and debilitated liver functions. Moreover, the microsomal cytochrome P450 system (CYP2E1) of the liver also involved in its metabolism contributes to the induction of alcoholic toxicit. 8
A study of the literature reveals that ethanol-induced oxidative stress contributes towards the etiology of alcohol-associated hepatic ailments by increasing the proinflammatory cytokine and chemokine expression, causing an increase in macrophage and neutrophil infiltration at the site of injury resulting in inflammation, necrosis, and apoptosis of hepatocytes.9,10 Long-term ethanol exposure activates the microsomal cytochrome P450 system and potentiates CYP2E1 activity, which plays an essential part in reactive oxygen species (ROS) generation. These ROS molecules damage constituents of cells and disturb the balance between the generation of oxidants and their clearance through anti-oxidant protection system leading to oxidative stress and induction of lipid peroxidation.11–14 The oxidation of mitochondrial proteins, lipids, and proteins triggers necrosis and apoptosis of hepatocytes enhancing disease progression and liver damage. 15 In addition, several inflammatory cascades are triggered by oxidative stress, secondary to tissue damage, leading to massive cytokines production. Nuclear factor-kappa B (NF-κB), a transcription factor and its related pathways act as a central player in the proinflammatory cascade. 16 The alcohol-induced liver disease triggers enhanced NF-κB signaling that in turn stimulates the expression of various other cytokines resulting in the inflammatory response.17,18 Kupffer cell stimulation induced by ethanol promotes the expression of a range of proinflammatory cytokines for instance tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6) which are implicated in ethanol-induced liver injury.19,20 TNF-α, the most important mediator of the mammalian proinflammatory response, has been documented to bring about apoptosis of hepatocytes and hepatic injury via cathepsin B-mediated pathway. 21 Furthermore, IL-6 results in significant kupffer cell injuries through its reduced products, ADH, and ALDH activities due to chronic and excessive consumption of ethanol, resulting in severe liver cell damage. 22
Currently, naturally occurring compounds have gained tremendous research interest to mitigate and prevent diseases in humans. Flavonoids are natural compounds having widely distributed throughout the plant kingdom as secondary metabolites. One of the most extensively studied and well-recognized compounds is a plant-derived bioactive flavonol, myricetin that, has immense nutraceutical value. The flavonoid myricetin (3, 5, 7-trihydroxy-two-(3, 4, 5-trihydroxyphenyl) -4H-chrome-4-1, also known as arbutin, is a phenolic compound that is present abundantly in various fruits, berries, tea, nuts, red wine, vegetables, and various medicinal herbs.23,24 Myricetin performs many critical biochemical and pharmacological functions, including potent anti-oxidant, anti-hypertensive, analgesic, antitumor, epigenetic modulation, antidiabetic, and anti-allergic anti-microbial, anti-inflammatory, and cardio/neuroprotective effects.25,26 In addition, myricetin has the ability to control major cellular enzymatic functions. In foods containing oils and fats, myricetin is routinely used as a preservative to extend their shelf life, owing to its ability to prevent the oxidation of lipids. Numerous research studies to date have reported that myricetin plays a beneficial part in the protection against some degenerative diseases through scavenging free radicals, chelating metal ions, and enhancing endogenous defense system. 27 Myricetin exhibits its antioxidant function through oxidation of three classes of hydroxyl groups (3′, 4′, 5′-position) bonded to its B ring, via interactions with various enzymes and receptors. 28 Myricetin has been reported to attenuate lipopolysaccharide (LPS) mediated cardiac injury through inhibition of the NF-κB pathway and inflammatory cytokines. 29 A large number of researchers have reported that compounds with antioxidant potential have a beneficial impact on ethanol-induced toxicity. Therefore, maintenance of hepatic antioxidant potential is believed to prevent the toxic effect of ethanol intake.30-35
Materials and Methods
Chemicals
Bovine serum albumin (BSA), ethylene diamine tetraacetate (EDTA), 1-chloro-2,4-dinitrobenzene (CDNB), diosmin, flavine adenine dinucleotide (FAD), reduced glutathione (GSH), oxidized glutathione (GSSG), thiobarbituric acid (TBA), xanthine, 2,6-dicholorophenolindophenol, and ascorbic acid were obtained from Sigma Aldrich, USA. All chemicals used in this study were of the highest available quality.
Experimental procedure
The current study assessed the effectiveness of myricetin in protecting ethanol-induced hepatic injuries. The total number of rats procured and included in the study was twenty four. These rats were arbitrarily categorized into four groups with six animals per group. Group-I animals were administered with vehicle (distilled water) and this group was labeled as control Group I. Group II, III and IV were treated orally with sequential (per week) increase in dose of ethanol (5, 8, 10, and 12 g/kg b wt per week in each group) for 28 days. The sequential increase in dose/concentration of ethanol overcomes the tolerance produced by ethanol consumed at same dose. Oral myricetin treatment was given to Group-III and IV animals at the respective doses of 25 mg/kg b wt. and 50 mg/kg b wt, one hour before ethanol administration. Animals were sacrificed after 28 days of ethanol administration through cervical displacement after being subjected to mild anesthetic treatment. The heart was punctured and the blood samples were collected for investigating different serological parameters. Samples of hepatic tissue were simultaneously obtained to study different biochemical parameters.
Preparation of post-mitochondrial supernatant, cytosolic, and microsomal fractions
The livers were taken out and subjected to ice-cold saline (0.85% sodium chloride) washing. Liver tissue homogenates (10%) were obtained in a buffer solution (10 mM Tris-HCl, 250 mM sucrose, pH 7.4), and nuclear debris was separated by centrifugation in Eltek refrigerated centrifuge for 10 min at 3000 rpm. The obtained aliquot was subjected to centrifugation for 20 min at 12,000 rpm to get PMS that served as the source of various enzymes. Ultra-centrifugation was carried out in the resulting supernatant for 1 h at 34,000 rpm to acquire a cytosolic fraction for assessing ADH activity - The obtained residue was washed with homogenizing buffer to attain microsomal fraction for CYP 2E1 activity. The whole process was done at 4°C.
Activity assessment of Catalase enzyme
Claiborne's method 36 was used to measure the activity of the catalase enzyme.
Assessment of lipid peroxidation
The lipid peroxidation (LPO) assay was performed by Wright et al. method. 37
Assessment of Glutathione (GSH)
The assessment of GSH was done with the protocol given by Jollow et al. 38
Activity assessment of Glutathione peroxidase (GPx)
GPx activity was evaluated using the method described by Mohandas et al. 39
Activity assessment of GR
Carlberg and Mannervik’s method 40 was used to determine the activity of GR.
Activity assessment of superoxide dismutase (SOD)
To measure the activity of SOD, the method given by Marklund & Marklund 41 was used.
The activity of CYP 2E1
The method of Reinke and Moyer 42 for estimating p-nitrophenol hydroxylation was used to analyze CPY2E1’s catalytic activity.
Activity assessment of ADH
Bonnichsen and Brink's method 43 was used to calculate the activity of ADH.
Activity assessment of serum ALT and AST
Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities were assessed by Reitman and Frankel’s method. 44
Activity assessment of lactate dehydrogenase
Korenberg's procedure 45 was used to determine the activity of serum lactate dehydrogenase.
Activity assessment of xanthine oxidase (XO)
The method given by Corte and Stirpe, 46 was employed in assessing the activity of XO.
Estimation of lipids
Commercially available kits (Cogent, Span diagnostics Ltd, India) were used to measure triglycerides (TGs), high density lipoproteins (HDL), low density lipoproteins (LDL), and total cholesterol (TC). The analysis was performed as per the guidelines offered by the manufacturer. 47
Estimation of Nitrites
Assay for estimation of nitrite was done by the method of Green et al., 48
Assessment of level of cytokines and transcription factor (TNF-α, IL-6, and NF-κB)
The cytokine/ transcription factor levels (TNF-α, IL-6, and NF-κB) were estimated with an enzyme-linked immunosorbent assay (ELISA) kit (eBioscience, Inc., San Diego, USA). The sample preparation was done in phosphate buffer saline (1xPBS, pH, 7.4) that contained a protease inhibitor cocktail. ELISA Plate Reader was used to perform the analysis as directed by the manufacturer.
Protein Estimation
Lowry et al. method 49 was used to measure the protein content in each sample with BSA taken as standard.
Results
Myricetin-induced effects on ethanol metabolizing enzymes
In ethanol-treated mice, the induction of CYP2E1 was substantially higher than in control animals (p<0.001). Myricetin treatment at both dosages drastically re-established CYP2E1 levels (p<0.05, p<0.01), respectively (Figure 1(a)). In comparison to the control group, the liver tissue of the ethanol-treated group revealed a considerable (p<0.001) rise in ADH activity. The ethanol-induced activity of ADH was significantly reduced by myricetin at both dosages (p<0.05, p<0.05) (Figure 1(b)). Myricetin mitigates ethanol metabolizing enzyme CYP 2E1 and ADH activity in the liver. (a) Ethanol administration has been found to elevate CYP 2E1 activity group II, (***p<0.001). Myricetin administration at both the doses significantly restores the ethanolinduced CYP 2E1 activity in group III (#p<0.05) group IV (##p<0.01). Each value represents mean ± SEM. n=6. (b) Ethanol administration in group-II caused a significant increase in ADH activity (***p<0.001) compared to control group-I. Treatment with myricetin markedly restored ADH activity in group-III (#p<0.05) and group-IV (#p<0.05) as compared to group-II. Each value represents mean ± SEM. n=6.
Myricetin-induced effects on TC and TGs
Result of treatment of myricetin on serum lipid profile in ethanol administered rats
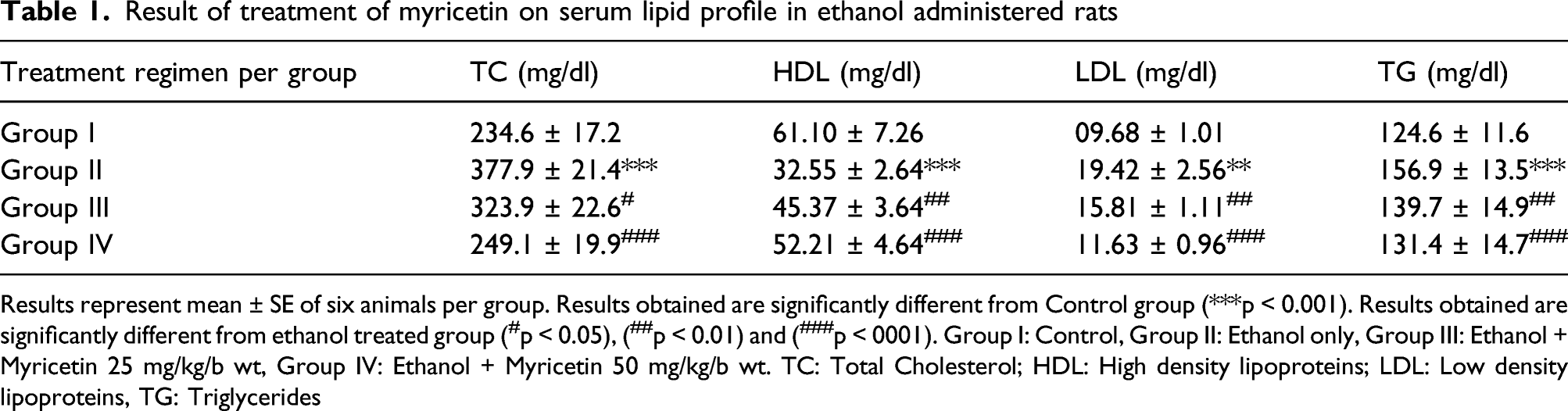
Results represent mean ± SE of six animals per group. Results obtained are significantly different from Control group (***p < 0.001). Results obtained are significantly different from ethanol treated group (#p < 0.05), (##p < 0.01) and (###p < 0001). Group I: Control, Group II: Ethanol only, Group III: Ethanol + Myricetin 25 mg/kg/b wt, Group IV: Ethanol + Myricetin 50 mg/kg/b wt. TC: Total Cholesterol; HDL: High density lipoproteins; LDL: Low density lipoproteins, TG: Triglycerides
Myricetin-induced effects on the activity of XO and lipid peroxidation (hepatic-membrane damage)
The activity of hepatic XO was drastically elevated in the ethanol-inoculated group (p<0.001) than in the control group. Both of the doses of myricetin observed significant recovery in the XO activity (p<0.01, p<0.001) (Figure 2(a)). The production of MDA was quantified for assessment of hepatic oxidative damage in ethanol-inoculated Wistar rats. Compared to the control group, a substantial increase in ethanol-induced MDA production (p<0.001) was found in the ethanol-treated group. Our findings show that myricetin treatment significantly reinstated the integrity of the membrane, as depicted by a decrease in hepatic MDA levels (p<0.01, p<0.001) (Figure 2(b)). Effect of myricetin and ethanol administration on XO activity and MDA levels in the liver. (a) Ethanol administration has been found to elevate XO activity significantly (group II, (***p<0.001). Myricetin administration at both doses significantly restores the ethanol-induce XO activity in group III (##p<0.01) group IV (###p<0.001). (b). Ethanol administration in group II caused a significant increase (***p<0.001) in MDA levels compared to control group-I. Treatment with myricetin significantly decreased MDA levels in group-III (##p<0.01) and group-IV (###p<0.001) as compared to group-II. Each value represents mean ± SEM. n=6.
Myricetin-induced restoration of hepatic antioxidant activities
Results of treatment of myricetin on antioxidant enzymes like GSH, GST, GR, XO, SOD, catalase and GPX on Ethanol induced redox imbalance.

Results represent mean ± SE of six animals per group. Results obtained are significantly different from Control group (***p < 0.001) Results obtained are significantly different from ethanol treated group (#p < 0.05), (##p < 0.01) and (###p < 0001). Group I: Control, Group II: Ethanol only, Group III: Ethanol + Myricetin 25 mg/kg/b wt, Group IV: Ethanol + Myricetin 50 mg/kg/b wt
Myricetin-alleviated hepato-toxicity induced by ethanol
In groups treated with ethanol, a remarkable elevation was seen in serum levels of hepato-toxicity markers including ALT, LDH, and AST (P<0.001) than the control group (Figure 3(a)–(c)). Myricetin treatment significantly normalized all of the serum toxicity indicators, that is, ALT (p<0.05, p<0.001) (Figure 3(a)), LDH (p<0.01, p<0.001) (Figure 3(b)), and AST (p<0.05, p<0.001) (Figure 3(c)). Effect of myricetin and ethanol administration on liver toxicity marker ALT, LDH, and AST. (a) Ethanol administration in group-II caused a significant increase (***p<0.001) in the liver toxicity marker ALT level compared to control group-I. Treatment with myricetin significantly decreased level of ALT in group- III (#p<0.05) and group-IV (###p<0.001) as compared to group-II. Each value represents mean ± SEM. n=6. (b) Ethanol administration led to the cellular toxicity and resulted in the increase of LDH in group-II (***p<0.001) as compared to group I. Treatment with myricetin significantly decreased cytotoxicity in group III (##p<0.01) and group IV (###p<0.001) as compared to group-II. Each value represents mean ± SEM. n=6. (c) Ethanol administration in group-II caused a significant increase (***p<0.001) in the level of liver toxicity marker AST compared to control group-I. Treatment with myricetin significantly decreased the level of AST in group-III (#p<0.05) and group-IV (###p<0.001) as compared to group-II. Each value represents mean ± SEM. n=6.
Myricetin restored inflammatory biomarkers (NF-κB, IL-6, and TNF-α)
The levels of inflammatory biomarkers (NF-κB, IL-6, and TNF-α) in the serum of ethanol administered to rats were considerably (p<0.001) elevated than the control group. At both treatment doses, myricetin remarkably restored NF-κB level (p<0.05, p<0.001) (Figure 4(a)), TNFα level (p<0.01, p<0.001) (Figure 4(b)), and IL6 (p<0.05, p<0.001) (Figure 4(c)). Effect of myricetin and ethanol administration on some cytokines NF-κB levels. (a) Ethanol administration in group-II caused significant increase (***p<0.001) in NF-κB levels as compared to control group-I. Treatment with myricetin significantly decreased NF-κB levels in group-III (#p<0.05) and group-IV (###p<0.001) as compared to group-II. (b) Ethanol administration in group-II caused significant increase (***p<0.001) in TNF-α levels as compared to control group-I. Treatment with myricetin significantly decreased TNF-α levels in group-III (##p<0.01) and group-IV (###p<0.001) as compared to group-II. (c) Ethanol administration in group-II caused significant increase (***p<0.001) in IL-6 levels as compared to control group-I. Treatment with myricetin significantly decreased IL-6 levels in group-III (#p<0.05) and group-IV (###p<0.001) as compared to group-II. Each value represents mean ± SEM. n=6.
Myricetin reduced nitrite levels induced by ethanol
The groups administered with ethanol demonstrated a substantial rise in the levels of nitrites (p<0.001) in comparison to the control group (Figure 5). Both treatment doses of myricetin effectively restored the normal levels of nitrites (p<0.01, p<0.001) (Figure 5). Effect of myricetin and ethanol administration on nitrite levels. In ethanol administered group-II, the nitrite levels were significantly increased (***p<0.001) as compared to control group-I. Treatment with myricetin significantly attenuated nitrite levels in group III (#p<0.05) and group IV (##p<0.01) as compared to group-II. Each value represents mean ± SEM. n=6.
Myricetin prevented the pathological changes in hepatic tissue
The microscopic analysis of liver tissue slices from various treatment groups demonstrated evident alterations in comparison to liver tissue of the control group (Figure 6). Rats treated with ethanol exhibited a marked vacuolar degeneration and prominent necrosis around the central vein. Furthermore, as demonstrated by inflammatory cell infiltration, ethanol elicited an inflammatory reaction in the hepatic tissue around the central vein. On the other hand, both treatment doses of myricetin were shown to protect the hepatic histology from changes induced by ethanol (Figure 6). Effect of myricetin on ethanol induced histological changes in liver tissue of rats: Liver section of rat control group (group I) showing normal architecture. Ethanol-treated group showing inflammatory response around central vein with vacuolar degeneration and necrosis. Myricetin administration at both the doses (group-II and group-III) reveals lesser vacuolar degeneration and decreased inflammatory response around the central vein.
Discussion
Alcohol-associated disorders at present are one of the most taxing health problems linked to social and economic consequences. Alcohol consumption continues to persist as one of the most common and important reasons for chronic hepatic disorders across the globe. 50 Alcohol intake can disrupt the delicate equilibrium of the body’s pro and anti-oxidant mechanisms, resulting in oxidative stress. The generation of reactive oxygen metabolites, for instance hydroxyl radical (OH·), hydrogen peroxide (H2O2), and superoxide (O2-·) in alcohol metabolism is the basic reason for alcohol-induced hepatic damage. 51 ADH and CYP2E1 are the crucial enzymes that convert alcohol to cytotoxic acetaldehyde associated with dioxygen reduction into several ROS. 52 Administration of myricetin significantly reduced the activities of these enzymes, thereby suppressing ROS-induced tissue damage caused by alcohol. Oxidation of alcohol by CYP2E1 and ADH also results in the production of NADH 53 which consequentially enhances the activity of XO. 54 The production of NADH is linked to the consumption of NADPH which subdues the GR-induced decrease of GSSG and consequently exhausts the content of GSH and other enzymes dependent on glutathione. GSSG suppresses cellular defense mechanisms and also enhances alcohol-associated oxidative stress and injury. 52 Activation of XO leads to the generation of superoxide radicals using acetaldehyde as a substrate. 55 Myricetin administration significantly reduced XO activity and helped restore hepatic glutathione in ethanol-administered rats.
Additionally, antioxidant enzymes such as SOD and CAT have a prominent role in safeguarding cells from the damage caused by free radicals.55-56 Administration of myricetin exhibited a marked increase in SOD and CAT activities in ethanol-administered rats. Furthermore, MDA is considered a biomarker for the evaluation of lipid peroxidation since it is generated by oxidation and degeneration of membrane lipids. 57 Therefore, the present study indicates the potential of myricetin as a hepatoprotective agent owing to its ability to maintain glutathione-dependent enzymatic systems and control free radical-mediated lipid peroxidation.
Alcohol stimulates the expression of inflammatory cytokines including, IL-6, IL-1β, and TNF-α. These cytokines further augment the generation of oxidants in the parenchyma of hepatic tissue and Kupffer cells resulting in their death due to necrosis.57-59 In the current experiment, alcohol administration elevated inflammatory markers (TNF-α, IL-1β, NF-κB), leading to necrosis and injury of hepatic tissue. Moreover, myricetin administration was seen to attenuate these pro-inflammatory cytokines and markers of cell necrosis. 60 The present findings show that myricetin impedes the production of alcohol-mediated inflammatory cytokines, thereby preventing cytokine-mediated hepatic damage.
A significant correlation exists between cellular damage and enzyme secretion. 60 Levels of lactate dehydrogenase (LDH), blood urea nitrogen (BUN), alanine aminotransferase (ALT), aspartate aminotransferase (AST), and creatinine in serum are the reliable indicators for assessing the hepatic function. 61 Results of the current study show that chronic administration of alcohol leads to a rise in the indices of hepatic dysfunction (AST, LDH, ALT, creatinine, and BUN) which concur with the published references.62,63 The rise in the activity of these serum biomarkers can be attributed to the escape of cytosolic enzymes into the circulation owing to damage of hepatic parenchyma, disruption in the enzyme synthesis machinery, and variation in membrane permeability. 64 Administration of myricetin led to restoration of ALT, AST, BUN, LDH, and creatinine levels in serum of ethanol-administered animals. This possibly occurs by maintaining the integrity of the hepato-cellular membranes, thereby indicating the potential hepatoprotective effect of myricetin.
Alcohol, on the one hand, stimulates biosynthesis while decreasing the catabolism of both cholesterol and fatty acids, leading to their buildup in the liver, consequently giving rise to hyperlipidemia. Alcohol increases the synthesis of cholesterol by increasing the activity of β-hydroxy- methyl-glutaryl CoA (HMG-CoA) reductase, the rate-determining enzyme of cholesterol biosynthesis. 65 Chronic alcohol administration also leads to microsomal induction that accelerates the oxidation of alcohol and enhances the production of triacylglycerols.62,63,66 Myricetin administration reduces cholesterol as well as triglyceride levels. Reduced cholesterol levels may be attributed to reduced absorption from the intestine and increased catabolism or decreased synthesis. The diminished levels of triglycerides may be caused by reduced synthesis of free fatty acids, increased breakdown, or reduced glycerol formation. 65
Alcohol intake increases the level of nitric oxide (NO). It might lead to toxicity via peroxynitrite, a powerful oxidant that can cause tyrosine residue nitration and inactivation of numerous biologically imperative proteins and enzymes.67,68 Following the previous studies, treatment with myricetin reduce nitrites in alcohol-administered rats. 69 Hence, myricetin treatment can potentially prevent reactive nitrogen species (RNS)-mediated damage to vital macromolecules (for instance, proteins, DNA, and lipids) of the hepatocytes which may occur due to alcohol consumption.
In summary, the current study depicts that alcohol causes hepatotoxicity due to generation of free radicals, disruption of redox potential, NF-κB stimulation, lipid peroxidation, and release of proinflammatory cytokines (TNF-α, IL-1β). These pathways lead to cellular necrosis and disruption of hepatic parenchyma. This study points to the therapeutic potential of myricetin in reducing alcohol-induced hepatic injury through modulation in the metabolism of alcohol, alleviation of oxidative stress, and suppression of inflammation. Hence myricetin may present a peculiar protective agent against alcohol-induced hepatic injury.
Footnotes
Acknowledgements
The authors acknowledge and extend their appreciation to the Researhers Supporting Project Number (RSP-2021/124) King Saud University, Riyadh, Saudi Arabia. The authors are thankful to the Faculty of Veterinary Science and Animal Husbandry, SKUAST-Kashmir, J&K, India, for providing the necessary facilities to complete a significant part of this project.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Researchers Supporting Project number (RSP-2021/124), King Saud University, Riyadh, Saudi Arabia.