
Abstract
Background
Atractylenolide III (ATL III) is a natural bioactive compound, that possesses anti-inflammatory, antioxidant, and neuroprotective properties. However, whether ATL III can protect against neuronal injury induced by cerebral ischemia/reperfusion (I/R) have not yet been studied. This study aimed to investigate the protective effects of ATL III on neuronal injury using an oxygen-glucose deprivation/reperfusion (OGD/R) model in HT22 cells.
Methods
Establishment of OGD/R model to induce HT22 cell injury in vitro. Cell viability, live-dead cell staining, oxidative stress levels, and pro-inflammatory cytokine levels were detected using kits. Cell apoptosis was observed by flow cytometry, and the expression of Bax, Bcl-2, and Caspase-3 proteins was detected by western blot.
Results
ATL III significantly alleviates OGD/R-induced cell injury, as evidenced by the increased cell viability and reduced apoptosis rate. ATL III increased the levels of superoxide dismutase (SOD) and glutathione (GSH), while reducing malondialdehyde (MDA), reactive oxygen species (ROS), and the levels of TNF-α, IL-1β, and IL-6. The protein expression of Bax and Caspase-3 was downregulated, while Bcl-2 expression was upregulated by ATL III.
Conclusion
ATL III as a potential therapeutic agent for reducing neuronal injury by mitigating oxidative stress, apoptosis, and inflammation.
Keywords
Introduction
Stroke is mainly classified into hemorrhagic stroke (HS) and ischemic stroke (IS), with IS accounted for approximately 87% of all strokes.1–3 Currently, intravenous thrombolysis is the primary therapeutic strategy for IS, however, the restoration of cerebral blood flow often triggers cerebral ischemia-reperfusion (I/R) injury, which exacerbates neurological deficits, making IS a leading cause of mortality and long-term disability worldwide.4,5 Therefore, there is an urgent need for novel neuroprotective agents that can mitigate the deleterious effects of I/R injury.
The pathological process of I/R injury is complex and multifactorial, involving oxidative stress, apoptosis, excitotoxicity, and inflammation, all of which contribute to neuronal death and subsequent neurological deficits. 6 Reactive oxygen species (ROS) play a central role in this process, not only cause oxidative stress but also trigger inflammatory cascades, exacerbate the neuronal injury and apoptosis.7,8 Both antioxidants and anti-inflammatory drugs effectively alleviate the neuronal damage caused by I/R.9–12 Therefore, alleviating oxidative stress and suppressing the inflammatory response represent a critical therapeutic strategy for reducing I/R-induced neuronal injury.
Atractylenolide III (ATL III), a bioactive sesquiterpene lactone isolated from Atractylodes macrocephala Koidz, Codonopsis pilosula (Franch.) Nannf
The present study is the first to evaluate the protective effect of ATL III against oxygen-glucose deprivation/reperfusion (OGD/R)-induced injury in HT22 hippocampal neurons, a well-established in vitro model of I/R injury was selected. Additionally, we also preliminary explored its underlying mechanism based on reducing oxidative stress, apoptosis, and inflammation, thereby offering new insights into its potential as a therapeutic agent for IS.
Materials and methods
Chemicals and reagents
ATL III (purity>98%, HPLC) was purchased from Yuanye Bio-Technology (Shanghai, China). Edaravone (EDA, purity>99%, HPLC) was purchased from Sigma-Aldrich (St Louis, MO, USA). Dulbecco modified Eagle medium (DMEM), penicillin-streptomycin solution, fetal bovine serum (FBS), and EDTA-free trypsin were purchased from HyClone (Logan, UT, USA). Glucose-free DMEM (Gibco, Waltham, MA, USA). Cell counting kit-8
Cell culture
HT22 mouse hippocampal neurons were obtained from Procell (CL-0697, Wuhan, China). Cells were cultured in DMEM containing 100 U/mL streptomycin and penicillin, and supplemented with 10% FBS at 37°C and 5% CO2.
OGD/R model
Cells were seeded in plates at a density of 8×105 cells/mL, when cells grow to 60%–70% density, the medium was replaced with glucose-free DMEM and incubated in an anoxic chamber with 5% CO2 and 95% N2 at 37°C for 10 h. Then, cells were restored to a normal glucose-containing DMEM with 5% CO2 and 95% O2 for another 6 h. The time of hypoxia and reoxygenation was based on our preliminary experiment. ATL III and EDA were added during the reoxygenation period. The experimental schedule is displayed in Figure 1(a). ATL III alleviates cell injury induced by OGD/R in HT22 cells. HT22 cells were treated with OGD for 10 h and treated with different concentrations of ATL III or EDA for 6 h during reoxygenation period. (a) The experimental schedule. (b) Cytotoxicity of ATL III on HT22 cells. (c) The effects of ATL III on cell viability in OGD/R cells. (d) Cell morphology under light microscope. (e) Calcein AM/PI staining. Green represents live cells, and red represents dead cells. (f) The proportion of dead cells. n = 6 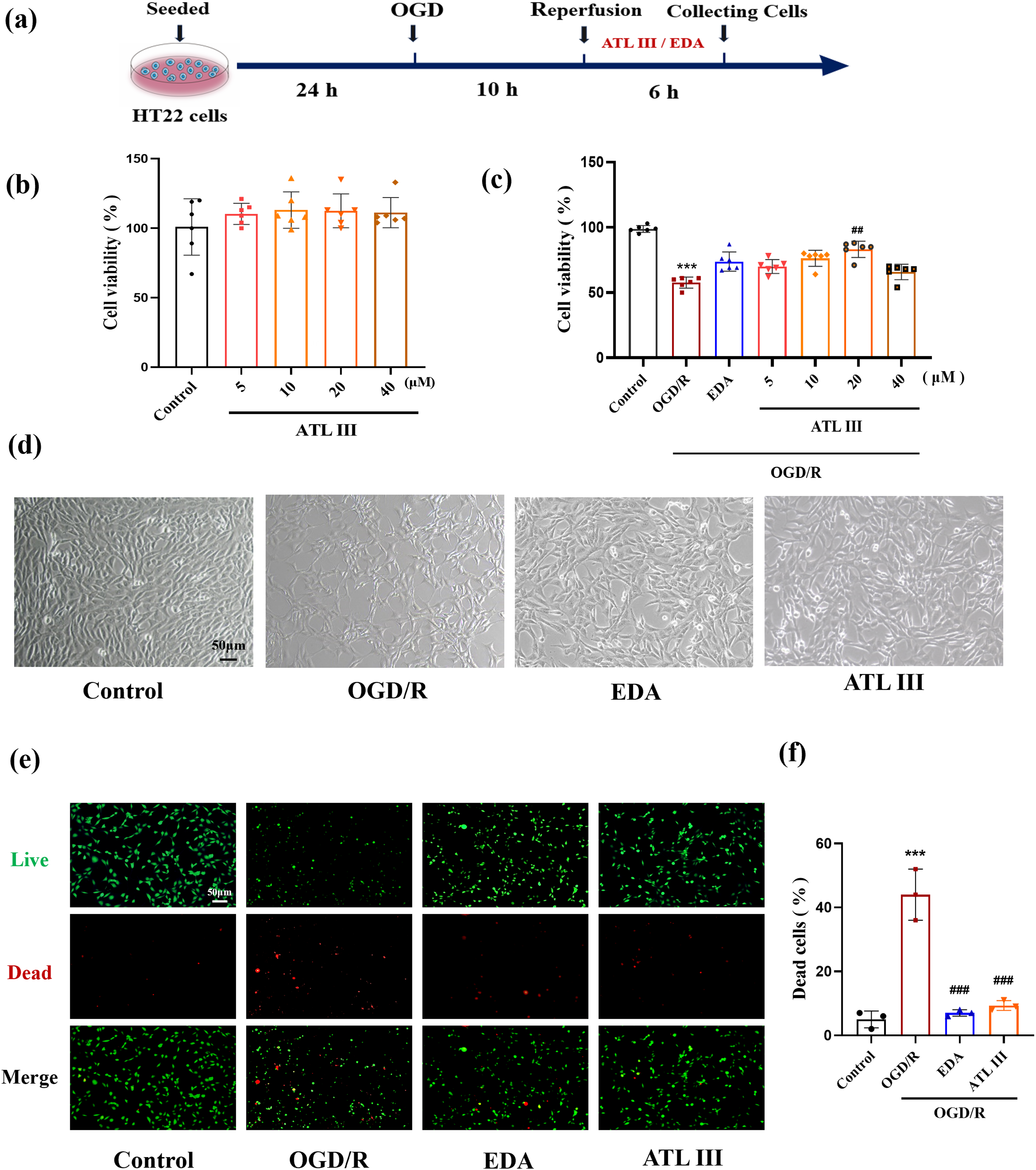
CCK-8 assay
HT22 cells were seeded in 96-well plates (5×104 cells/mL) and cultured for 24 h. Then, cells were subjected by OGD/R modeling and treated with ATL III (5, 10, 20, and 40 μM) and EDA (50 μM). 10 µL of CCK-8 reagent was added and incubated for 30 min in the dark. The optical density (OD) was determined at 450 nm by a microplate reader (Biotek, VT, USA).
Calcein AM/PI staining
HT22 cells were seeded in 24-well plates (1.5×105 cells/mL) and cultured for 24 h. Then, cells were subjected by OGD/R modeling and treated with ATL III (20 μM) and EDA (50 μM). Liver and dead cells were stained using a Calcein-AM/PI staining kit, and captured by an inverted fluorescence microscopy (Leica, Switzerland, Germany).
ROS detection
The ROS level was detected using a DCFH-DA staining kit. Briefly, HT22 cells were seeded in 6-well plates (3×105 cells/mL) and cultured for 24 h. Then, cells were subjected by OGD/R modeling and treated with ATL III (20 μM) and EDA (50 μM). 1 mL of DCFH-DA was added and incubated at 37°C for 25 min in the dark. The cells were washed three times with PBS and captured by inverted fluorescence microscopy (Ex: 488 nm; Em: 525 nm).
Determination of MDA, SOD, and GSH levels
HT22 cells were seeded in 60-mm dish (5×105 cells/mL) for 24 h. Then, cells were subjected by OGD/R modeling and treated with ATL III (20 μM) and EDA (50 μM). After collecting cells by centrifugation at 1000 rpm for 10 min, cells were lysed in RIPA solution by an ultrasonic pulverizer (300 W, ultrasonic for 5 sec, interval for 10 sec, 30 cycles). After centrifugating at 12,000 g for 20 min, the supernatants were used for the determination of MDA, SOD, and GSH levels according to the instruction of kits. The total protein concentration was measured using a BCA protein assay kit.
Determination of TNF-α, IL-1β, and IL-6 levels
After OGD/R modeling and treatment, cells were collected and lysed in RIPA solution by an ultrasonic pulverizer as described above. After centrifugating at 12,000 g for 20 min, the supernatants were used for the determination of TNF-α, IL-1β, and IL-6 levels using ELISA kits according to the instructions of the manufacturer.
Cell apoptosis detection
Cell apoptosis was detected using an Annexin V-FITC/PI double staining kit. Briefly, cells were subjected by OGD/R modeling and treated with ATL III (20 μM) and EDA (50 μM). 5 μL of Annexin V-FITC solution and 5 μL of PI solution was added and incubated for 5 min. Cell apoptosis was detected by a flow cytometry (CytoFLEX S, Beckman, CA, USA). The total apoptosis rate (%) = early apoptosis rate (%) + late apoptosis rate (%).
Western blotting
Total protein was extracted by centrifugation at 12,000 g for 20 min at 4°C. The protein concentration was measured by BCA protein assay kit. Proteins were separated by SDS-PAGE and transferred onto PVDF membranes (Millipore, MA, USA). The membrane was blocked in 5% skimmed milk for 2 h and incubated with primary antibodies for Caspase-3 (1:4,000), Bcl-2 (1:500), and Bax (1:2,000) overnight at 4°C. After washing three times, the membrane was incubated with goat anti-rabbit HRP-conjugated IgG (1:10,000) at room temperature for 1 h. Protein bands were reacted with ECL reagents and β-actin (1:30,000) was used as an internal reference. Image J software (National Institutes of Health, Bethesda, MD, USA) was used to quantify the integrated optical density (IOD).
Statistical analysis
Data were analyzed using SPSS 13.0 (SPSS, Chicago, IL, USA) statistical software and figures were drawn using GraphPad prism 5.0 (GraphPad, San Diego, CA, USA). Data were presented as mean ± standard deviation, and analyzed using one-way ANOVA with Tukey’s post hoc test. Differences with p < .05 were defined as significant.
Result
ATL III alleviates cells injury induced by OGD/R in HT22 cells
ATL III did not affect the cell viability at a concentration range from 5 to 40 μM (Figure 1(b)). Cell viability was decreased to 57% in the OGD/R group compared to the control group (p < .001), while 5, 10, and 20 μM ATL III increased cell viability to 69.7%, 73.7%, and 80.6%, respectively. In particular, 20 μM ATL III significantly increased cell viability (p < .01), which is better than 50 μM EDA (Figure 1(c)). The cell morphology under the light field was displayed in Figure 1(d). In addition, live-dead cell staining showed that 20 μM ATL III (p < .001) and EDA (p < .001) significantly reduced the proportion of dead cells compared to the OGD/R group (Figure 1(e) and (f)). These results demonstrated that ATL III alleviated cell injury induced by OGD/R in HT22 cells.
ATL III reduces OGD/R-induced oxidative stress in HT22 cells
Oxidative stress is one of the main causes of OGD/R-induced neuron injury.
19
Therefore, we first investigated the effects of ATL III on oxidative stress levels. As shown in Figure 2, a significant increase in ROS and MDA levels, and a reduction in GSH level and SOD activity were observed in the OGD/R group when comparing to the control group (p < .001). ATL III and EDA significantly reduced ROS and MDA levels, while increased GSH level and SOD activity (p < .001). These results indicated that ATL III reduced oxidative stress levels induced by OGD/R. ATL III alleviates OGD/R-induced oxidative stress in HT22 cells. HT22 cells were treated with OGD for 10 h and treated with different concentrations of ATL III or EDA for 6 h during reoxygenation period. (a) Cellular ROS was examined by DCFH-DA staining. (b) The quantitation of ROS level. (c) Cellular MDA levels. (d) Cellular SOD activity. (e) Cellular GSH level.

ATL III suppresses inflammation response induced by OGD/R in HT22 cells
Inflammation plays a critical role in the neuronal injury caused by ischemia and reperfusion.
20
Therefore, we investigated the effect of ATL III on pro-inflammatory cytokine levels in OGD/R-induced HT22 cells. As shown in Figure 3, the levels of IL-1β, IL-6, and TNF-α were significantly increased in the OGD/R group compared to the control group (p < .001), but dramatically decreased by ATL III (p < .001) and EDA (p < .001; p < .05; p < .05). ATL III reduces cytokine levels in OGD/R-induced HT22 cells. (a) The level of IL-1β. (b) The level of IL-6. (c) The level of TNF-α. n = 3.
***
p < .001 vs. control group;

ATL III reduces OGD/R-induced apoptosis in HT22 cells
The apoptosis rate was significantly increased in the OGD/R group (p < .001) and reduced in the ATL III (p < .001) and EDA groups (p < .001) (Figure 4(a)–(b)). In addition, the protein expression of Bax (p < .01) and Caspase-3 (p < .05) were increased and Bcl-2 (p < .05) was decreased in the OGD/R group compared to the control group. ATL III significantly reversed these changes, up-regulated the expression of Bcl-2 (p < .01), and down-regulated Bax (p < .001) and Caspase-3 expression (p < .01). EDA also reversed these protein expressions (p < .01) (Figure 4(c)–(f)). ATL III reduces cell apoptosis induced by OGD/R in HT22 cells. (a) Cell apoptosis detected by flow cytometry. (b) The quantitation of apoptosis rate. (c) The immunoblot bands of Bax, Bcl-2, and Caspase-3. (d) The relative quantification of Bax protein. (e) The relative quantification of Bcl-2 protein. (f) The relative quantification of Caspase-3 protein. n = 3.

Discussion
The present study demonstrated that ATL III exerts significant protective effects against OGD/R-induced injury in HT22 cells. ATL III not only improved cell viability but also reduced oxidative stress, inflammation, and apoptosis, which were key pathological processes involved in cerebral I/R injury, highlighting its potential as a promising therapeutic agent for the treatment of cerebral I/R injury.
Apoptosis is a critical factor underlying neuronal loss during I/R injury,21,22 and its inhibition is vital for reducing cerebral damage and promoting recovery. 23 In this study, our results demonstrated that ATL III significantly improved cell viability, with the best effect at 20 μM, surpassing even EDA, a widely recognized neuroprotective agent. This improvement in cell viability was further verified by live-dead cell staining, indicating the potent protective effect of ATL III against OGD/R-induced injury. In addition, ATL III markedly reduced OGD/R-induced cell apoptosis, as evidenced by decreased Bax and Caspase-3 levels, and increased Bcl-2 expression. Caspase-3, a key executioner caspase, facilitates the final steps of apoptosis, leading to DNA fragmentation and cell death. 24 Overactivation of Caspase-3 can result in excessive apoptosis, contributing to various neurological diseases, including Alzheimer’s disease and cerebral ischemia.25,26 Bax, a pro-apoptotic member of the Bcl-2 family, promotes apoptosis by permeabilizing the mitochondrial membrane, releasing cytochrome c, and activating Caspase-3. In contrast, Bcl-2 inhibits apoptosis by antagonizing Bax. 27 Previous research has shown that ATL III exerts neuroprotective effects by modulating apoptosis-related proteins. 28 Our findings demonstrated that ATL III downregulated Bax and Caspase-3 while upregulating Bcl-2, effectively reducing neuronal apoptosis induced by OGD/R, which is critical for preserving neuronal survival and function after ischemic injury.
Inflammation plays a pivotal role in the pathogenesis of I/R injury, 29 characterized by the release of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α. This neuroinflammatory response not only exacerbates brain damage but also impairs neurological recovery, highlighting the importance of anti-inflammatory in therapeutic strategies for I/R injury. 30 ATL III has been shown to suppress the production of pro-inflammatory cytokines and inhibit NF-κB pathway activation. 31 Additionally, ATL III reduced microglial activation and oxidative stress, thereby mitigating inflammation-induced neuronal damage. 18 In our study, ATL III treatment significantly reduced the elevated levels of pro-inflammatory cytokines in OGD/R-induced HT22 cells. This suppression of cytokines demonstrated the potential ATL III as a promising anti-inflammatory agent for IS therapy.
Oxidative stress is another key contributor to neuronal injury following I/R, primarily due to the overproduction of ROS and the depletion of endogenous antioxidant defenses such as SOD and GSH. 32 Accumulating oxygen free radicals trigger lipid peroxidation and MDA production, leading to DNA damage and apoptosis. In our study, OGD/R-induced HT22 cells exhibited elevated ROS and MDA levels, along with reduced GSH levels and SOD activity. ATL III treatment significantly reduced ROS and MDA levels while restored GSH and SOD activity, effectively mitigated oxidative stress, which is a critical step in preserving neuronal integrity and function during I/R injury. 33 Previous studies have shown that ATL III mitigates oxidative stress by activating the Nrf2/NQO1/HO-1 pathway, a crucial pathway for cellular defense against oxidative damage. 34 Our findings indicated that ATL III increased antioxidant enzyme levels, thereby reducing ROS and protecting cells from OGD/R-induced oxidative injury.
In summary, this study is the first to demonstrated that ATL III protects against OGD/R-induced injury in HT22 cells by reducing oxidative stress, inflammation, and apoptosis. These findings supported ATL III as a promising candidate for therapeutic strategies aimed at reducing neuronal damage and enhancing recovery following cerebral I/R injury. However, this study only focused on the protective effects of ATL III in an in vitro model, further research is necessary to validate these findings in vivo and to explore the detailed molecular pathways involved. Additionally, understanding the pharmacokinetics and optimal dosing of ATL III in the context of IS treatment are essential for its potential translation into clinical practice.
Footnotes
Acknowledgment
We thank all participators for their help.
Author contributions
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China [No. 82304771], Science and Technology Research and Development Project of Henan Province [No.232301420018].
Ethical statement
Data availability statement
All data presented in the present study are available on request from the corresponding author.