
Abstract
Paraoxonase 2 (PON2) is a powerful antioxidant that mediates cell survival under oxidative stress; however, its protection neurons against cerebral ischemia-reperfusion injury-induced oxidative stress remains unclear. This study aimed to determine the precise regulating role of PON2 in neuronal survival under oxidative stress. An in vitro model of cerebral ischemia-reperfusion injury was used to assess the effect of PON2 on oxidative stress induced by oxygen–glucose deprivation/reoxygenation (OGD/R). Results showed that PON2 expression in neurons was decreased due to OGD/R exposure. A series of functional experiments revealed that upregulated PON2 improved OGD/R-impaired viability and attenuated OGD/R-induced increases in apoptosis and reactive oxygen species in neurons. Decreased PON2 expression enhanced neuronal sensitivity to OGD/R-induced injury. Overexpressed PON2 markedly enhanced the expression of nuclear factor erythroid 2-related factor 2 (Nrf2) in the nucleus and increased the levels of Nrf2-mediated transcriptional activity. Furthermore, PON2 enhanced the Nrf2 activation by modulating glycogen synthase kinase-3β (GSK-3β). Inhibition of GSK-3β substantially abrogated the PON2 knockdown-mediated suppression of Nrf2 activity. Notably, Nrf2 inhibition partially reversed the neuroprotective effects of PON2 overexpression in OGD/R-exposed neurons. These findings indicate that PON2 alleviates OGD/R-induced apoptosis and oxidative stress in neurons by potentiating Nrf2 activation via GSK-3β modulation. This study highlights the potential neuroprotective function of PON2 against cerebral ischemia-reperfusion injury.
Introduction
Ischemic stroke is a major cause of death and permanent disability and generates substantial family and social economic burdens. 1 Brain ischemia is mainly caused by the sudden occlusion of the middle cerebral artery by a thrombus or embolism. 2 Intravenous thrombolytic therapy has been widely used to treat ischemic stroke; 3 however, rapid blood reperfusion could result in brain damage 4 referred to as cerebral ischemia-reperfusion injury. 5 The precise molecular pathogenesis of this condition is highly complicated, and one of its major causes is the excessive generation of reactive oxygen species (ROS) and subsequent oxidative stress. 6 Therefore, understanding the molecular mechanisms of oxidative stress induced by cerebral ischemia-reperfusion injury may facilitate the development of novel treatments for this disease.
Paraoxonase 2 (PON2) is a member of the paraoxonase family with a pivotal role in a wide range of biological processes. 7 This protein is ubiquitously expressed in various tissues and cell types 8 –10 and has no esterase activity but acts as a potent lactonase. 11 PON2 dysregulation has been implicated in a large number of diseases, including cancers, diabetes, and coronary artery diseases. 12 –14 A growing body of evidence suggests that this protein functions as a powerful antioxidant. 9,15,16 PON2 can retard atherosclerosis progression by slowing foam cell formation through oxidative stress reduction. 17 –19 Furthermore, the antioxidant capacity of PON2 has a protective effect against Pseudomonas aeruginosa infection. 20 PON2 knockdown increases ROS production in the kidneys and intestines. 16,21 This protein can also protect against acute myocardial ischemia-reperfusion injury by reducing oxidative stress. 22 These findings indicate that PON2 is closely related to oxidative stress-associated pathological processes and suggest its potential as a therapeutic target for treating oxidative stress-related diseases.
Normal functioning cells have low ROS concentrations. However, excessive ROS generation under harmful stress conditions can produce a state of oxidative stress within the cell and cause cell damage and apoptosis. 23 Nuclear factor erythroid 2-related factor 2 (Nrf2) is a redox-sensitive transcription factor that regulates a set of antioxidant and detoxifying genes and plays a key role in the cellular response to oxidative stress. 24 In response to oxidative stress, cytoplastic Nrf2 proteins are translocated into the nucleus where they bind to antioxidant response element (ARE) sequences in the promoter regions of genes and initiate the transcription of antioxidant- and detoxification-related genes. 25 Nrf2/ARE activation is controlled by various factors, including glycogen synthase kinase-3β (GSK-3β). 26 It has been reported that GSK-3β phosphorylates Nrf2 and promotes Nrf2 degradation, 27,28 which is important for the regulation of Nrf2 activity. In vitro and in vivo studies involving cerebral ischemia-reperfusion injury have revealed that GSK-3β inhibition enhances Nrf2 activity and produces neuroprotective effects. 29 –32 Upregulation of Nrf2 target genes, including heme oxygenase-1 (HO-1) and NAD(P)H quinone oxidoreductase-1 (NQO-1), protects against cerebral ischemia-reperfusion injury. 33,34 Therefore, elucidating the GSK-3β/Nrf2 axis in cerebral ischemia-reperfusion injury will improve our understanding of the molecular pathogenesis of this disease.
Recent studies have indicated that PON2 is profoundly neuroprotective, 35,36 but its protective function against cerebral ischemia-reperfusion injury remains unclear. This study aimed to determine the precise regulating role of PON2 in neuron survival under oxidative stress induced by oxygen-glucose deprivation/reoxygenation (OGD/R) by using an in vitro model of cerebral ischemia-reperfusion injury. Results showed that PON2 expression is reduced in the neurons exposed to OGD/R. A series of functional experiments revealed that PON2 upregulation remarkably improved the OGD/R-reduced viability and attenuated the OGD/R-induced increases in apoptosis and ROS accumulation in neurons. Further investigation suggested that PON2 overexpression enhanced Nrf2 activity via GSK-3β modulation. GSK-3β inhibition or Nrf2 activation partially reversed the PON2 knockdown-mediated increase in OGD/R-induced neuronal injury, whereas Nrf2 inhibition markedly diminished the PON2 overexpression-mediated neuroprotective effects. In summary, PON2 alleviates OGD/R-induced apoptosis and oxidative stress in neurons by potentiating Nrf2 activation through GSK-3β modulation.
Materials and methods
Neuronal HT22 cell culture
Mouse neuronal HT22 cells were provided by BeNa Culture Collection (Kunshan, Jiangsu, China) and grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco; Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (FBS) plus glutamine and sodium pyruvate. Cells were cultivated at 37°C in a humidified atmosphere containing 5% CO2.
In vitro model of OGD/R
HT22 neurons were washed with phosphate-buffered saline (PBS), placed in glucose-free DMEM, and exposed to hypoxic conditions (a mixture of 3% O2/5% CO2/92% N2) for 8 h. Thereafter, the medium was replaced by a complete medium containing glucose, and the cells were cultivated for either 12 or 24 h and allowed to re-oxygenate. The HT22 neurons cultured under normal conditions in complete medium during the experimental period were used as controls.
Real-time quantitative RCR (RT-qPCR) analysis
Total RNA was exacted and purified using RNeasy Mini Kit (Qiagen, Hilden, Germany) in accordance with the manufacturer’s instructions and then subjected to reverse transcription using PrimeScript RT master mix to synthesize cDNA templates (Takara Biomedical Technology, Beijing, China). The cDNA template was amplified with TB Green Fast qPCR mix (Takara Biomedical Technology) using standard thermal cycler parameters as follows: 95°C for 30 s, followed by 40 cycles of 95°C for 5 s and 60°C for 15 s. Data were analyzed using the 2−ΔΔCt method, and relative gene expression was calculated using β-actin as an internal control.
Western blot assay
HT22 neurons were homogenized in RIPA lysis buffer to facilitate total protein extraction. Nuclear proteins were extracted using a nuclear protein extraction kit (Beyotime, Shanghai, China). Protein concentrations were determined using a Bichinonic Acid (BCA) Assay Kit (Beyotime). Equal quantities of protein from each group were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis. Proteins were then electroblotted onto a polyvinylidene fluoride (PVDF) membrane and blocked with 5% non-fat milk in tris-buffered saline containing Tween 20 (TBST). The PVDF membrane was then incubated overnight at 4°C with primary antibodies, then washed with TBST, and probed using horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. The membrane was washed with TBST and immersed within an ECL chemiluminescent substrate solution to develop immunoreactive bands. Primary antibodies used in this study were as follows: anti-PON2 antibody (Abcam, Cambridge, UK), anti-β-actin antibody (Abcam), anti-Nrf2 antibody (Abcam), anti-histone H3 antibody (Abcam), and anti-histone H3 (Cell Signaling Technology, Danvers, MA, USA).
Cell transfection
The coding sequences (CDS) of PON2 were subcloned into pcDNA3.1 vectors to construct PON2 expression vectors. The specific siRNA targeting PON2 and control siRNA were synthesized by GenePharma (Shanghai, China). Constructed vectors or synthesized siRNAs were transfected into HT22 neurons by using Lipofectamine 3000 Reagent (Invitrogen; Thermo Fisher Scientific, Waltham, MA, USA) in accordance with the manufacturer’s specifications.
Cell viability assay
HT22 neurons were seeded into 96-well plates and cultured overnight under normal conditions to allow adherence to plates. After transfection and incubation, these neurons were subjected to OGD/R treatment. For viability measurement, 10 µl of cell counting kit-8 (CCK-8) reagent (Beyotime) was added to each well. After being cultured for 2 h at 37°C under normal conditions, the cells were detected by measuring their optical density (OD) at 450 nm by using a microplate reader (Bio-Rad, Hercules, CA, USA).
Cell apoptosis assay
HT22 neurons were collected and assessed using a commercial terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labeling (TUNEL) assay kit (RiBoBio, Guangzhou, China). These neurons were washed with PBS and fixed by adding 4% paraformaldehyde for 15 min at room temperature. The cells were rinsed with deionized water, permeabilized via incubation with 0.5% Triton X-100 for 10 min at room temperature, washed, and incubated with terminal transferase (TdT) buffer for 5 min at room temperature. TdT buffer was removed, and TdT enzyme incubation solution was added to the cells that were subsequently incubated for 2 h at 37°C in the dark. The reaction was ceased by adding stop reaction buffer solution, and the cells were washed with PBS. TUNEL-positive cells were observed and counted using a fluorescent microscope (Leica Microsystems, Heidelberg, Germany) with an excitation wavelength of 488 nm and an emission wavelength of 515 nm.
ROS assay
Intracellular ROS levels in HT22 neurons were assessed using a ROS Assay Kit (Beyotime) containing 2′,7′-dichlorofluorescein diacetate (DCFH-DA). For ROS detection, HT22 neurons were incubated with DCFH-DA (10 μM) in serum-free media for 30 min at 37°C in the dark, washed with serum-free medium, and subjected to flow cytometry to determine their fluorescence intensities.
Nrf2 luciferase reporter assay
Nrf2 transcriptional activity was monitored using a Nrf2 luciferase reporter vector pARE-luc (Beyotime) containing an ARE site. HT22 neurons were co-transfected with pARE-luc vectors, control Renilla Luciferase vectors, pRL-SV40, and PON2 expression vectors, or PON2 siRNA. The cells were then incubated for 48 h. After OGD/R treatment, luciferase activity was measured using the dual luciferase reporter gene assay kit (Beyotime) in accordance with the manufacturer’s instructions.
Statistical analysis
GraphPad Prism 8 software (GraphPad Software, San Diego, CA, USA) was used to process and analyze experimental data. All data were presented as mean ± standard deviation. Comparisons among multiple groups were conducted by using one-way ANOVA, followed by the Tukey’s post hoc test. Statistical significance was set at p < 0.05.
Results
PON2 expression in HT22 neurons was decreased after OGD/R exposure
To assess the role of PON2 in regulation of OGD/R-induced neuronal injury, we first examined whether level of PON2 varied in response to OGD/R exposure in HT22 neurons. OGD/R exposure resulted in a significant decrease in PON2 expression at mRNA and protein levels in HT22 neurons (Figure 1(a) to (c)). These data imply that PON2 expression is decreased when HT22 neurons are exposed to OGD/R.

Effect of OGD/R on PON2 expression in HT22 neurons. HT22 neurons were cultivated under OGD conditions for 8 h and then re-oxygenated in complete medium for either 12 or 24 h. (A) Effect of OGD/R on PON2 mRNA expression was measured via RT-qPCR. (B) Effect of OGD/R on PON2 protein levels was determined via Western blot. *p < 0.05 and **p < 0.01.
PON2 upregulation alleviated OGD/R-induced apoptosis and oxidative stress in HT22 neurons
To explore the precise function of PON2 in the regulation of OGD/R-induced neuronal injury, we detected the effects of enhanced PON2 activity in HT22 neurons. The HT22 cells transfected with PON2 expression vectors significantly upregulated the PON2 expression (Figure 2(a) to (c)). CCK-8 assay revealed that PON2 overexpression restored HT22 neuronal viability, which was impaired by OGD/R exposure (Figure 2(d)). Moreover, PON2 overexpression significantly decreased the OGD/R-induced apoptosis (Figure 2(e) and (f)) and ROS production (Figure 2(g) and (h)) in HT22 neurons. These data indicate that PON2 protects cells against OGD/R-induced neuronal injury.

Overexpression of PON2 decreases levels of OGD/R-induced apoptosis and oxidative stress in HT22 neurons. HT22 neurons were transfected with PON2 expression vectors or empty vectors (EV) for 48 h prior to OGD/R treatment. (a) PON2 mRNA expression was assessed via RT-qPCR and (b) and (c) PON2 protein expression was determined via Western blot. (D) Effect of PON2 overexpression on the viability of cells exposed to OGD/R was measured using a CCK-8 assay. (e) and (f) Effect of PON2 overexpression on OGD/R-induced apoptosis was assessed via TUNEL apoptosis assay. (g) and (h) Effect of PON2 overexpression on OGD/R-induced ROS generation was monitored using a reactive oxygen species assay. **p < 0.01 and ***p < 0.001.
PON2 loss enhanced the sensitivity of HT22 neurons to OGD/R-induced injury
To confirm that PON2 protects OGD/R-exposed neurons, we assessed the effects of PON2 loss-of-function. Transfection with PON2 siRNA markedly depleted the PON2 expression in the neurons exposed to OGD/R (Figure 3(a) and (b)). Functional experiments revealed that PON2 depletion further decreased the viability of OGD/R-exposed HT22 neurons (Figure 3(c)) and exacerbated the OGD/R-induced neuronal apoptosis (Figure 3(d) and (e)) and ROS production (Figure 3(f) and (g)). These findings suggest that PON2 depletion enhances the sensitivity of HT22 neurons to OGD/R-induced injury.
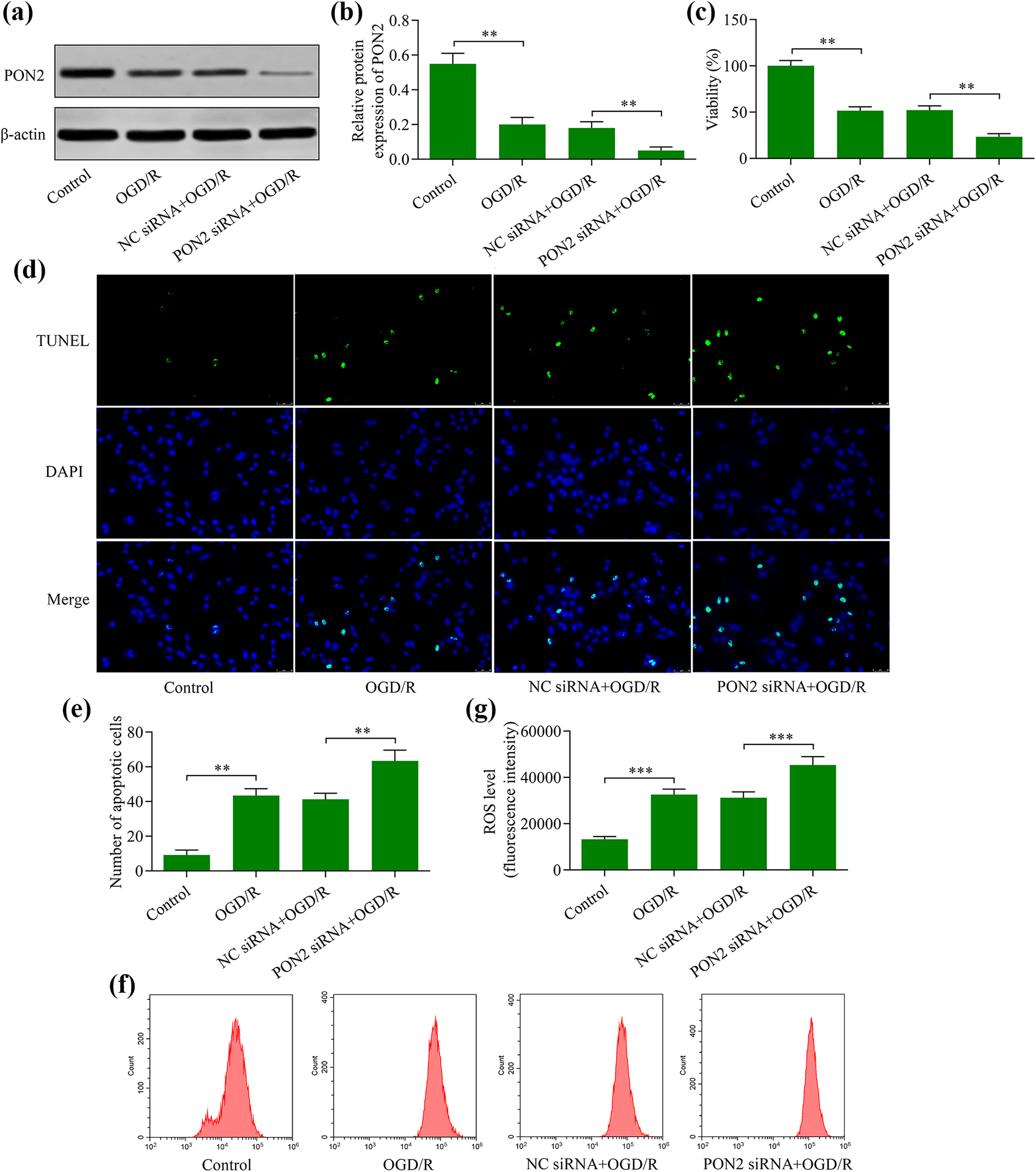
Knockdown of PON2 produces increased levels of OGD/R-induced neuronal injury. (a) and (b) PON2 protein expression assessed via Western blot HT22 in neurons transfected with either PON2 siRNA or negative control (NC) siRNA for 48 h prior to OGD/R treatment. (c) Effect of PON2 knockdown on OGD/R-stressed cells was measured using a CCK-8 assay. (d) and (e) Effect of PON2 knockdown on OGD/R-induced apoptosis was assessed using a TUNEL apoptosis assay. (f) and (g) Effect of PON2 knockdown on OGD/R-induced ROS generation was monitored using a ROS assay. **p < 0.01 and ***p < 0.001.
PON2 overexpression activated Nrf2
To elucidate molecular mechanism in which PON2 protects neuronal cells from OGD/R-induced injury, we next investigated the effects of PON2 overexpression on the activity of Nrf2, a master regulator of antioxidant defense. PON2 overexpression markedly enhanced the nuclear expression of Nrf2 (Figure 4(a) and (b)) and upregulated the transcriptional activity of Nrf2/ARE (Figure 4(c)). In addition, PON2 overexpression significantly enhanced the expression of Nrf2 target genes including HO-1 and NQO-1 (Figure 4(d) and (e)). These data suggest that PON2 positively regulates Nrf2 activity.

PON2 overexpression potentiates Nrf2 activation. (a) and (b) Effect of PON2 overexpression on Nrf2 nuclear expression was determined via Western blot. Histone H3 served as the loading control. (c) Effect of PON2 overexpression on Nrf2/ARE transcriptional activity was monitored using a luciferase reporter assay. Effect of PON2 overexpression on the expression of (d) HO-1 and (e) NQO-1 was measured via RT-qPCR. *p < 0.05 and **p < 0.01.
PON2 enhanced Nrf2 activation via GSK-3β modulation
We next investigated the regulatory effect of PON2 on GSK-3β, a key regulator of Nrf2. The data revealed that GSK-3β phosphorylation was enhanced by PON2 overexpression (Figure 5(a) and (b)), but decreased by the knockdown of this protein (Figure 5(c) and (d)). Since low levels of GSK-3β phosphorylation are indicative of its hyperactivation, we examined the effect of GSK-3β inhibition on PON2 knockdown-mediated Nrf2 activity. PON2 knockdown attenuated Nrf2 activation, which was markedly reversed GSK-3β inhibition (Figure 5(e)). Moreover, the PON2 knockdown-induced exacerbation of OGD/R-induced apoptosis and ROS production was markedly abrogated by GSK-3β inhibition (Figure 5(f) and (g)). These findings suggest that PON2 enhances Nrf2 activity by modulating GSK-3β.
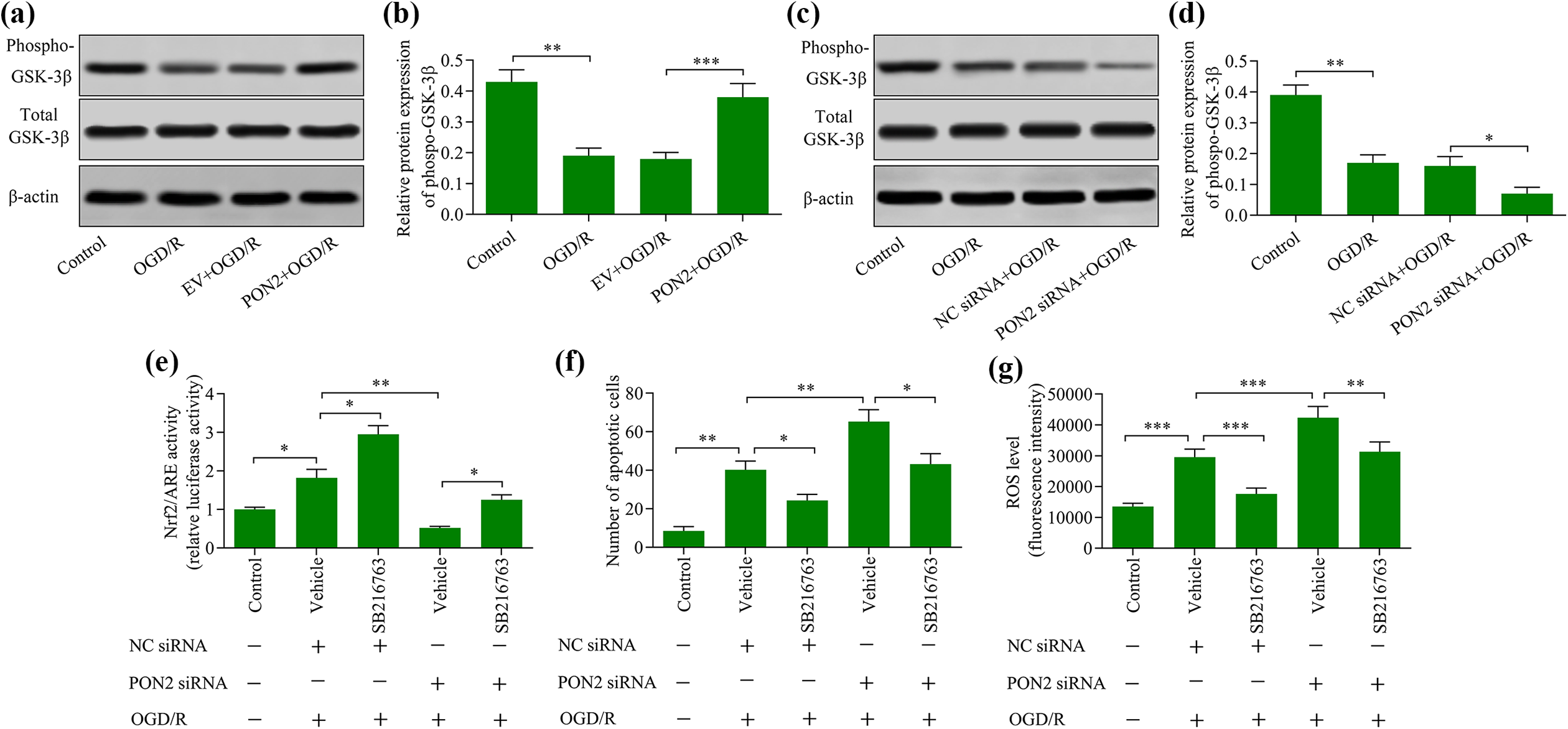
GSK-3β inhibition reversed PON2 knockdown-mediated effects of OGD/R exposure in neurons. Effect of (a) and (b) PON2 overexpression or (c) and (d) PON2 knockdown on levels of GSK-3β phosphorylation was examined using Western blot. HT22 neurons were transfected with PON2 siRNA and incubated for 48 h in the presence of SB216763, a GSK-3β inhibitor, prior to OGD/R exposure. (e) Effect of GSK-3β inhibition on Nrf2 activation was determined using a luciferase reporter assay. (f) Effect of GSK-3β inhibition on levels of apoptosis was assessed using a TUNEL apoptosis assay. (g) Effect of GSK-3β inhibition on ROS production was determined using a reactive oxygen species assay. *p < 0.05, **p < 0.01, and ***p < 0.001.
Nrf2 inhibition abolished the neuroprotective effects mediated by PON2 overexpression
To confirm whether Nrf2 contributes to PON2-mediated neuroprotection, we next evaluated the effects of Nrf2 inhibition on PON2 overexpression-induced inhibition of OGD/R-induced apoptosis and ROS production. Treatment with a Nrf2 inhibitor markedly reversed the Nrf2 activation in HT22 neurons in OGD/R-exposed neurons transfected with or without PON2 vectors (Figure 6(a)). The PON2 overexpression-induced suppression of OGD/R-induced apoptosis and ROS production were remarkably reversed by Nrf2 inhibition (Figure 6(b) and (c)). In addition, we assessed the effects of Nrf2 activation on PON2 knockdown-enhanced OGD/R injury. The data showed that treatment with a Nrf2 activator markedly upregulated the Nrf2 activation in OGD/R-exposed neurons transfected with or without PON2 siRNA (Figure 6(d)). Moreover, Nrf2 activation significantly abrogated the PON2 knockdown-mediated enhancement of OGD/R-induced apoptosis and ROS production (Figure 6(e) and (f)). These results show that PON2 protects cells from OGD/R-induced neuronal injury by enhancing Nrf2 activity.

PON2 protects HT22 cells from OGD/R-induced neuronal injury through enhancement of Nrf2 activation. HT22 neurons were transfected with PON2 expression vectors and were cultivated for 48 h in the presence of 2 3M ML385, a Nrf2 inhibitor, prior to OGD/R exposure. (a) Effect of Nrf2 inhibition on Nrf2 transcriptional activity was determined using a luciferase reporter assay. (b) Effect of Nrf2 inhibition on levels of cellular apoptosis was measured using a TUNEL apoptosis assay. (c) Effect of Nrf2 inhibition on ROS generation was assessed using a reactive oxygen species assay. HT22 neurons were transfected with PON2 siRNA and incubated for 48 h in the presence of tBHQ, a Nrf2 activator, prior to OGD/R exposure. (d) Effect of Nrf2 activation on Nrf2 transcriptional activity was determined using a luciferase reporter assay. (e) Effect of Nrf2 activation on cellular apoptosis was measured using a TUNEL apoptosis assay. (f) Effect of Nrf2 activation on ROS generation was assessed using a reactive oxygen species assay. *p < 0.05, **p < 0.01, and ***p < 0.001.
Discussion
This study found that PON2 mediates the protection against OGD/R-induced injury in HT22 neurons. Our data revealed that PON2 upregulation markedly attenuated the OGD/R-induced apoptosis and ROS generation and promoted neuronal survival after OGD/R exposure. Moreover, this study elucidates that the neuroprotective effects of PON2 are related to its enhancement of Nrf2 activity via GSK-3β modulation. These findings indicate that PON2 acts as a novel and key regulator of Nrf2 and underscore that the PON2/GSK-3β/Nrf2 axis may be involved in neuronal protection against OGD/R-induced apoptosis and oxidative stress (Figure 7).
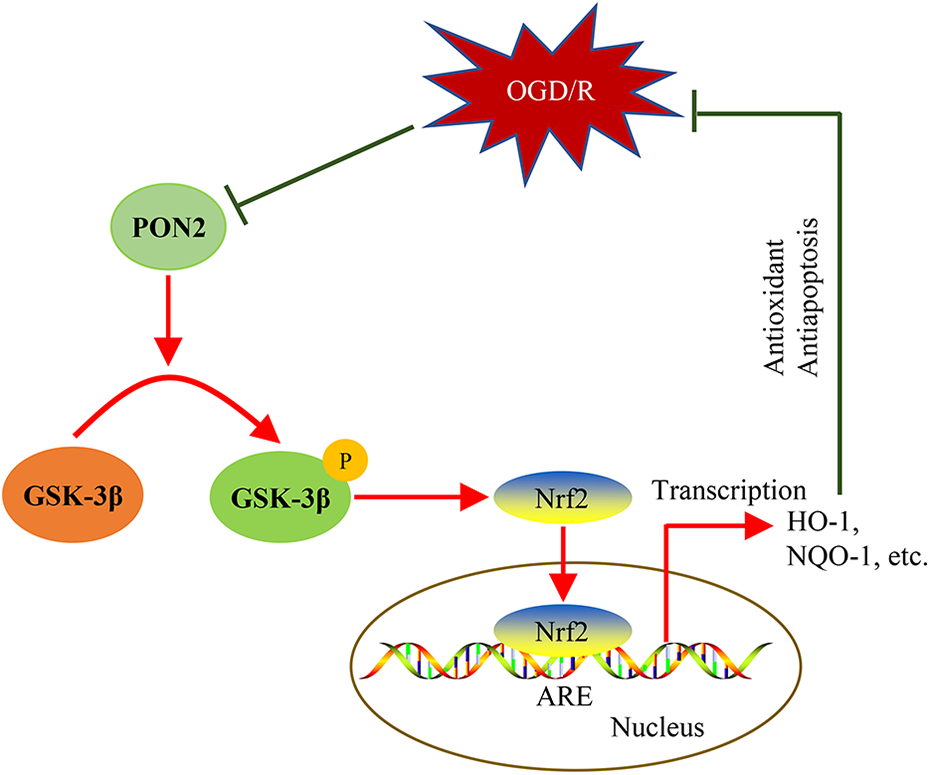
Graphical model depicting the potential molecular mechanism of PON2-mediated protection against OGD/R-induced apoptosis and oxidative stress.
PON2 is an anti-apoptotic protein that enhances cell survival under adverse conditions. 37 Upregulation of PON2 protects cancer cells from irradiation-induced apoptosis. 38 In PON2-dificient macrophages, the proapoptotic pathway is overactivated in response to endoplasmic reticulum stress. 39 The capacity of PON2 to promote survival is closely related to its antioxidant capacity. 40 PON2 overexpression suppresses ROS production and enables vascular cells to survive endoplasmic reticulum stress. 15 Considering that the excessive ROS generation induced by ischemia-reperfusion injury is a major cause of cellular apoptosis, we hypothesized that PON2 may protect against ischemia-reperfusion injury. A recent study reported that PON2 knockout exacerbates acute myocardial ischemia-reperfusion injury by upregulating apoptosis and ROS production in cardiomyocytes. 22 Moreover, PON2 overexpression alleviates hypoxia-induced apoptosis and ROS generation in cardiomyocytes in vitro. 22 However, whether PON2 is involved in cerebral ischemia-reperfusion injury remains largely unknown. In the present work, PON2 was found to protect neurons from OGD/R-induced apoptosis and ROS production. The findings confirm that PON2 exerts a neuroprotective effect that is associated with its anti-apoptotic and antioxidant capacities. Furthermore, this protein may regulate oxidative stress in cerebral ischemia-reperfusion injury.
The neuroprotective role of PON2 has been highlighted. 35 This protein is widely expressed in neurons of all brain regions, and its expression decreases with age in mice. 41 PON2-deficient neurons are highly sensitive to oxidants, such as hydrogen peroxide and 2,3-dimethoxy-1,4-naphthoquinone, oxidative stress, and cytotoxicity. 41,42 Induction of PON2 by quercetin contributes to quercetin-mediated antioxidant and neuroprotective effects. 43 It has been reported that PON2 interacts with DJ-1, a Parkinson’s disease-related gene that exerts neuroprotection against oxidative stress, 44 to protect neurons from 1-methyl-4-phenylpyridinium-induced apoptosis and oxidative stress. 45 These findings suggest that PON2 has a profound neuroprotective function. However, its protection against OGD/R-induced neuronal injury has not been well studied. This work revealed that PON2 also exerted neuroprotective effect in OGD/R-exposed neurons. Its upregulation markedly attenuated OGD/R-induced apoptosis and ROS production, whereas its knockdown enhanced the sensitivity of neurons to PGD/R-induced injury. The present findings and previous studies indicate that PON2 is an attractive therapeutic target for neuroprotection.
To date, the molecular mechanisms underlying PON2-mediated neuroprotective effects have been largely unknown. In this study, the regulatory effect of PON2 on Nrf2, a master regulator of antioxidant defense, was investigated. The results showed that PON2 upregulation markedly promoted the nuclear translocation of Nrf2 and increased the Nrf2-mediated gene transcription. Therefore, PON2 may exert its neuroprotective function by enhancing Nrf2 activation. Indeed, Nrf2 inhibition markedly abrogated the PON2-mediated neuroprotective effect in OGD/R-exposed neurons. In addition, our work elucidates that PON2 mediates Nrf2 activation by modulating GSK-3β, a key regulator of Nrf2 activation. 27,28 The regulatory effect of PON2 on GSK-3β phosphorylation has been previously reported. PON2 upregulation can attenuate radiation-induced apoptosis by inhibiting GSK-3β. 46 A recent work revealed that PON2 protects against myocardial ischemia-reperfusion injury by inactivating GSK-3β, which is associated with the upregulation of GSK-3β phosphorylation. 22 In accordance with these findings, the present data showed that GSK-3β phosphorylation was enhanced by PON2 overexpression but decreased by PON2 knockdown. Moreover, the treatment of cells with a GSK-3β inhibitor markedly reversed the OGD/R-induced neuronal injury that was exacerbated by PON2 knockdown. All these findings indicate that PON2 promotes Nrf2 activation by regulating GSK-3β, which modulates PON2-mediated neuroprotective effects in OGD/R-exposed neurons.
In summary, data presented here reveal that PON2 protects neurons from OGD/R-induced apoptosis and oxidative stress by enhancing Nrf2 activity via GSK-3β modulation. These findings suggest the critical role of PON2 in regulating Nrf2 activation and highlight the new mechanism in which PON2 alleviates oxidative stress. In addition, the PON2/GSK-3β/Nrf2 regulatory axis may play a role in the protection against cerebral ischemia-reperfusion injury-induced oxidative stress. However, further studies using animal models are warranted to validate the molecular mechanism in which PON2 regulates cerebral ischemia-reperfusion injury.
Footnotes
Authors’ contributions
JB designed the study, performed the experiments, and drafted the manuscript. PJ performed the experiments. YZ analyzed the data. KW analyzed the data. GW designed the study and reviewed the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.