
Abstract
Senescence marker protein 30 (SMP30) is a senescence marker molecule and identified as a calcium regulatory protein. Currently, SMP30 has emerged as a cytoprotective protein in a wide range of cell types. However, the role of SMP30 in regulating neuronal survival during cerebral ischemia/reperfusion injury remains unclear. In the present study, we aimed to investigate the biological function and regulatory mechanism of SMP30 on neuronal survival using a cellular model induced by oxygen-glucose deprivation/reoxygenation (OGD/R). The results showed that SMP30 expression was significantly decreased by OGD/R exposure in neurons. Functional experiments demonstrated that SMP30 overexpression significantly rescued the decreased cell viability and attenuated the apoptosis and reactive oxygen species generation in OGD/R-exposed neurons. By contrast, SMP30 knockdown exhibited the opposite effect. Mechanism research revealed that SMP30 overexpression contributed to the activation of nuclear factor erythroid 2-related factor (Nrf2)/antioxidant response element (ARE) signaling associated with downregulation of Kelch-like ECH-associated protein (Keap1). Keap1 overexpression or Nrf2 silencing significantly reversed SMP30-mediated neuroprotection against OGD/R-induced injury. Overall, these findings demonstrate that SMP30 overexpression protects neurons from OGD/R-induced apoptosis and oxidative stress by enhancing Nrf2/ARE antioxidant signaling via inhibition of Keap1. These data highlight the importance of the SMP30/Keap1/Nrf2/ARE signaling axis in regulating neuronal survival during cerebral ischemia/reperfusion injury.
Introduction
Ischemic stroke is an extremely severe disorder in the central nervous system accompanied with high morbidity and mortality worldwide. 1 Early reperfusion is the preferred treatment for cerebral ischemia. However, the reperfusion causes additional tissue damage, including neuronal apoptosis, blood-brain barrier destruction, and cerebral edema; this damage is termed cerebral ischemia/reperfusion injury. 2 Patients with cerebral ischemia/reperfusion injury have an extremely poor quality of life due to the permanent disability. Thus, this dysfunction is currently a considerable public health threat that causes a huge economic burden on society and the family. 1 Oxidative stress is a hallmark of cerebral ischemia/reperfusion injury. 3 Excessive production of reactive oxygen species (ROS) induced by cerebral ischemia/reperfusion injury contributes to brain injury through various signaling pathways. 4 Therefore, a better understanding of the mechanisms that underlie oxidative-stress-mediated neuronal injury is important in developing potential and effective therapies for cerebral ischemia/reperfusion injury treatment.
Senescence marker protein-30 (SMP30) is a novel aging-associated protein with a molecular weight 34 kDa, which plays an important role in regulating various biological functions. 5,6 SMP30 is abundantly expressed in a wide range of tissues, including brain, lung, liver, and kidney. 7 SMP30 is identified as a calcium regulatory protein that plays an important role in maintaining intracellular calcium homeostasis. 8 Interestingly, SMP30 expression in these tissues declines with age and SMP30 downregulation accelerates cell senescence. 9 SMP30 knockout mice have a short lifespan and lack ascorbic acid biosynthesis. 10 Moreover, SMP30 can protect cells against age-associated deterioration. 11 Indeed, accumulating evidence clearly indicates that SMP30 exerts a cytoprotective role against oxidative stress and cell apoptosis. 12 –15 Overexpression of SMP30 protects rat kidney epithelial cells from undergoing apoptosis, whereas SMP30 deficiency increases susceptibility to radiation-induced apoptosis in mouse small intestine and to tumor necrosis factor-α and Fas-induced apoptosis in the liver. 16 –18 Further, SMP30 exerts a critical role in regulating oxidative stress produced under various adverse stimulus. 19 –21
Nuclear factor erythroid 2-related factor (Nrf2) is a master regulator of cellular defense mechanisms against endogenous and exogenous stresses through the activation of multiple cytoprotective genes. 22 Nrf2 translocates to the nucleus in response to oxidative stress and binds to antioxidant response element (ARE) in the regulatory regions of many cytoprotective genes via dimerizing with small Maf proteins. 23 However, the activation of Nrf2/ARE signaling is regulated by various factors, such as Kelch-like ECH-associated protein 1 (Keap1). Keap1 is an E3 ubiquitin ligase substrate-recognition adaptor subunit that sequesters Nrf2 in the cytoplasm and promotes Nrf2 degradation via the proteasome. 24 Notably, Keap1/Nrf2 signaling plays a critical role in regulating oxidative stress during cerebral ischemia/reperfusion injury and has emerged as an attractive therapeutic target for this disease. 25
Intriguingly, SMP30 reportedly exerts cardioprotection in ischemia/reperfusion-induced injury of heart. 19 However, whether SMP30 is involved in cerebral ischemia/reperfusion injury remains undetermined. In this study, we aimed to explore the function and regulatory mechanism of SMP30 in oxygen-glucose deprivation/reoxygenation (OGD/R)-induced neuronal injury, an in vitro model for cerebral ischemia/reperfusion injury. We found that SMP30 expression was significantly decreased by OGD/R exposure in neurons. Functional experiments demonstrated that SMP30 deficiency further decreased the viability and exacerbated the apoptosis and ROS production in OGD/R-exposed neurons. By contrast, SMP30 overexpression alleviated OGD/R-induced neuronal injury. Mechanism research revealed that SMP30 overexpression enhanced the activation of Nrf2/ARE signaling associated with Keap1 downregulation. Keap1 overexpression or Nrf2 silencing significantly reversed SMP30-mediated neuroprotection against OGD/R-induced injury. Overall, these findings demonstrate that SMP30 overexpression protects neurons from OGD/R-induced injury by enhancing Nrf2/ARE antioxidant signaling via inhibition of Keap1. These data highlight the importance of the SMP30/Keap1/Nrf2/ARE signaling axis in regulating neuronal survival during cerebral ischemia/reperfusion injury.
Materials and methods
Cell culture
Mouse hippocampal HT22 neurons which have been widely used for the study of OGD/R-induced injury 26 –28 were purchased from BeNa Culture Collection (BNCC, Kunshan, Jiangsu, China). These cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS). Cells were maintained in a humidified incubator (95% air/5% CO2) and grown at 37°C.
Establishing a OGD/R-induced injury model in HT22 neurons
To mimic ischemic-like conditions in vitro, the HT22 neurons were cultured in DMEM without glucose and incubated under hypoxic conditions (5% CO2/3% O2/92% N2). After 8-h culture, the medium was replaced with DMEM that contained 4.5 g/l glucose. The cells were then cultured under normoxic conditions (95% air/5% CO2) for 24 h.
Oligonucleotide synthesis, plasmid construction, and transfection
Small interfering RNA (siRNA) sequences that targeted SMP30 or Nrf2 were synthesized by RiboBio (Guangzhou, China) and transfected into cells with the riboFECT CP Transfection Kit (RiboBio) according to the manufacturer’s guidelines. The complementary DNA (cDNA) sequences encoding the SMP30 or Keap1 opening reading frame were amplified by polymerase chain reaction (PCR) and subcloned into the pcDNA3.1 plasmid. The plasmids were transfected into cells with the Lipofectamine 3000 Reagent (Invitrogen; Thermo Fisher Scientific), as per the manufacturer’s protocols.
Real-time quantitative PCR (RT-qPCR) analysis
Total cellular RNA was isolated with TRIzol Reagent (Invitrogen) and reversely transcribed into cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems; Thermo Fisher Scientific), following the manufacturers’ standard protocols. The synthesized cDNA was used as the template for RT-qPCR using PowerUp SYBR Green Master Mix (Invitrogen) with appropriate primers. Gene amplification was performed with a Applied Biosystems 7500 Real-Time PCR System. The thermal cycling parameters were: 95°C for 2 min followed by 40 cycles of 95°C for 15 s and 60°C for 60 s. Relative gene expression was calculated according to the 2−ΔΔCt method; β-actin was used as the internal control. The details of primers were as follows: SMP30 (101 bp), forward: 5’-GAACTACAGGTGTGGGGAGTC-3’ (Tm: 61.5°C) and 5’-reverse: TGACCGTATCCCATCGACAAATA-3’ (Tm: 60.9°C); HO-1 (100 bp), forwards: 5’- AAGCCGAGAATGCTGAGTTCA-3’ (Tm: 61.7°C) and reverse: 5’- GCCGTGTAGATATGGTACAAGGA-3’ (Tm: 61.2°C); NQO-1 (144 bp), forward: 5’-AGGATGGGAGGTACTCGAATC-3’ (Tm: 60.2°C) and reverse: 5’-AGGCGTCCTTCCTTATATGCTA-3’ (Tm: 60.2°C); β-actin (154 bp), forwards: 5’- GGCTGTATTCCCCTCCATCG-3’ (Tm: 61.8°C) and reverse: 5’-CCAGTTGGTAACAATGCCATGT-3’ (Tm: 61.1°C).
Western blot analysis
Cells were lysed in Cell Lysis Buffer for Western Blot (Beyotime, Shanghai, China) containing protease and phosphatase inhibitor cocktail. The supernatants were harvested by centrifugation at 12000g, 4°C for 5 min. The protein centration in the collected supernatants was measured by the BCA Protein Assay Kit (Beyotime). Protein samples were added to loading buffer and boiled at 100°C for 5 min. Equivalent amounts of protein extracts were loaded onto sodium dodecyl sulfate polyacrylamide gels and resolved with electrophoresis. The separated proteins were then transferred to polyvinylidene difluoride (PVDF) membranes using the semi-dry method. The membranes were then incubated with 5% nonfat milk in Tris-buffered saline with Tween 20 (TBST) for 1 h at room temperature to block nonspecific binding. Thereafter, the membranes were incubated with anti-SMP30 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-Keap1 (Abcam, Cambridge, UK), anti-β-actin (Abcam), anti-Nrf2 (Abcam), or anti-lamin B2 (Abcam) primary antibodies overnight at 4°C. The membrane was washed with TBST three times and then probed with horseradish-peroxidase-linked secondary antibody for 1 h at room temperature. For protein visualization, membranes were immersed in the Pierce ECL Western Blotting Substrate (Thermo Scientific) in the dark. The gray value of protein bands was quantified using Image-Pro Plus 6.0.
CCK-8 cell viability assay
HT22 neurons were seeded into a 96-well plate at a density of 5 × 103 cells/well and cultured overnight prior to transfection. HT22 neurons were transfected with SMP30 siRNA or SMP30 expression vector for 48 h followed by OGD/R exposure. To determine cell viability, 10 μl/well CCK-8 solution were added to the HT22 neurons and incubated for 2 h at 37°C. The absorption values of the colorimetric solution at 450 nm were measured with a microplate reader (Bio-Tek Instruments, Winooski, VA, USA).
Flow cytometric detection of cell apoptosis
HT22 neurons were harvested after the indicated treatment and washed with phosphate-buffered saline (PBS). Cells (1 × 105) were resuspended in 195 µl Annexin V-FITC binding buffer, followed by addition of 5 µl Annexin V-FITC and 10 µl propidium iodide (Beyotime). Cells were mixed gently and incubated in the dark for 15 min at room temperature. Then, the samples were immediately analyzed by flow cytometry, and apoptotic cells were assessed with FlowJo software.
Caspase-3 activity assay
Caspase-3 activity, which indicates the apoptosis level, was measured by a colorimetric capase-3 assay kit (Abcam). According to the kit’s protocol, HT22 neurons were resuspended in the Cell Lysis Buffer and incubated for 10 min on ice. Then, the supernatants were collected by centrifugation and transferred to a new 96-well plate at 50 μl/well. Then, 5 µl of 4 mM DEVD-p-NA substrate and 50 µl Reaction Buffer were added to each well. HT22 neurons were gently mixed and then incubated for 2 h at 37°C. The absorbance at 405 nm was measured with a microplate reader.
Determination of ROS production
For determination of the ROS level, HT22 neurons were placed into serum-free DMEM that contained 10 μM DCFH-DA (Beyotime) followed by incubation for 20 min at 37°C. Thereafter, HT22 neurons were washed with serum-free medium and resuspended in PBS for cytometric analysis.
ARE luciferase reporter assay
HT22 neurons were cotransfected with the ARE-Luc reporter plasmid (Promega, Madison, WI, USA), Renilla luciferase plasmid and SMP30 expression plasmid and incubated for 48 h. HT22 neurons were then subjected to OGD/R exposure before detection. For detection of luciferase activity, cells were lysed and assayed with the Dual-Luciferase Reporter Assay System (Promega).
Statistical analysis
Experimental data are presented as mean ± standard deviation. Data were processed with GraphPad Prism 6 (Software Inc., San Diego, CA, USA), and statistical differences were determined by one-way analysis of variance (ANOVA) followed by Bonferroni’s post-hoc test. The criterion of significance between groups was set at p < 0.05.
Results
SMP30 loss exacerbated OGD/R-induced injury in HT22 neurons
To explore the potential role of SMP30 in regulating OGD/R-induced neuronal injury, we examined its expression level in HT22 neurons after OGD/R treatment. Both SMP30 mRNA and protein expression were significantly decreased in HT22 neurons following OGD/R exposure (Figure 1A and B). To elucidate the biological function of SMP30 in regulating OGD/R injury, we detected the effect of SMP30 depletion on OGD/R-induced apoptosis and ROS production in HT22 neurons. SMP30 expression was markedly depleted in HT22 neurons transfected with SMP30 siRNA (Figure 1A and B). OGD/R exposure significantly decreased HT22 neuron viability (Figure 1C). However, SMP30 depletion further decreased the viability of OGD/R-exposed HT22 neurons (Figure 1C). Moreover, OGD/R-induced apoptosis and caspase-3 activation were significantly augmented by SMP30 depletion (Figure 1D and E). Additionally, the increased ROS production in OGD/R-exposed HT22 neurons was enhanced by SMP30 depletion (Figure 1F). Overall, these data suggest that SMP30 deficiency exacerbates OGD/R-induced neuronal injury.
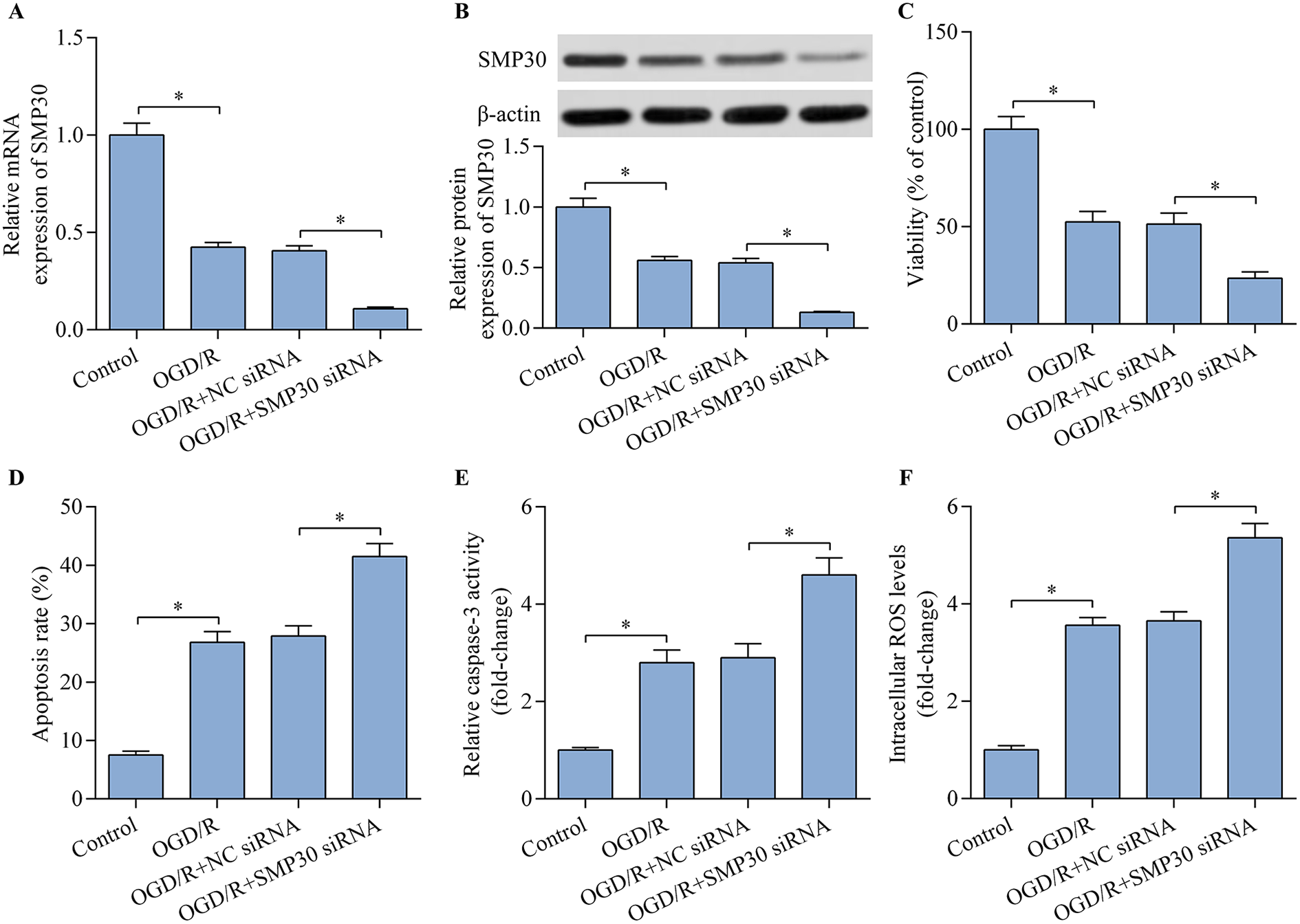
SMP30 knockdown exacerbated OGD/R-induced injury of HT22 neurons. Relative SMP30 (A) mRNA and (B) protein expression were detected by RT-qPCR and western blot, respectively. HT22 neurons were transfected with negative control (NC) siRNA or SMP30 siRNA for 48 h prior to OGD/R injury. (C) The effect of SMP30 knockdown on cell viability was assessed with the CCK-8 cell viability assay. The effect of SMP30 knockdown on cell apoptosis was evaluated by (D) Annexin V-FITC/PI flow cytometric analysis and (E) caspase-3 activity assay. (F) The effect of SMP30 knockdown on intracellular ROS level was monitored by flow cytometric analysis. *p < 0.05.
SMP30 overexpression protected HT22 neurons from OGD/R-induced injury
Considering that SMP30 depletion exacerbated OGD/R-induced neuronal injury, we hypothesized that SMP30 overexpression may exert neuroprotective effect. To test this hypothesis, we upregulated SMP30 expression in OGD/R-exposed neurons by transfecting the SMP30 expression vector (Figure 2A). As expected, SMP30 overexpression significantly rescued the decreased viability of OGD/R-exposed HT22 neurons (Figure 2B). Moreover, OGD/R-induced apoptosis and ROS production were markedly repressed by SMP30 overexpression (Figure 2C–E). Collectively, these results suggest a neuroprotective function for SMP30.
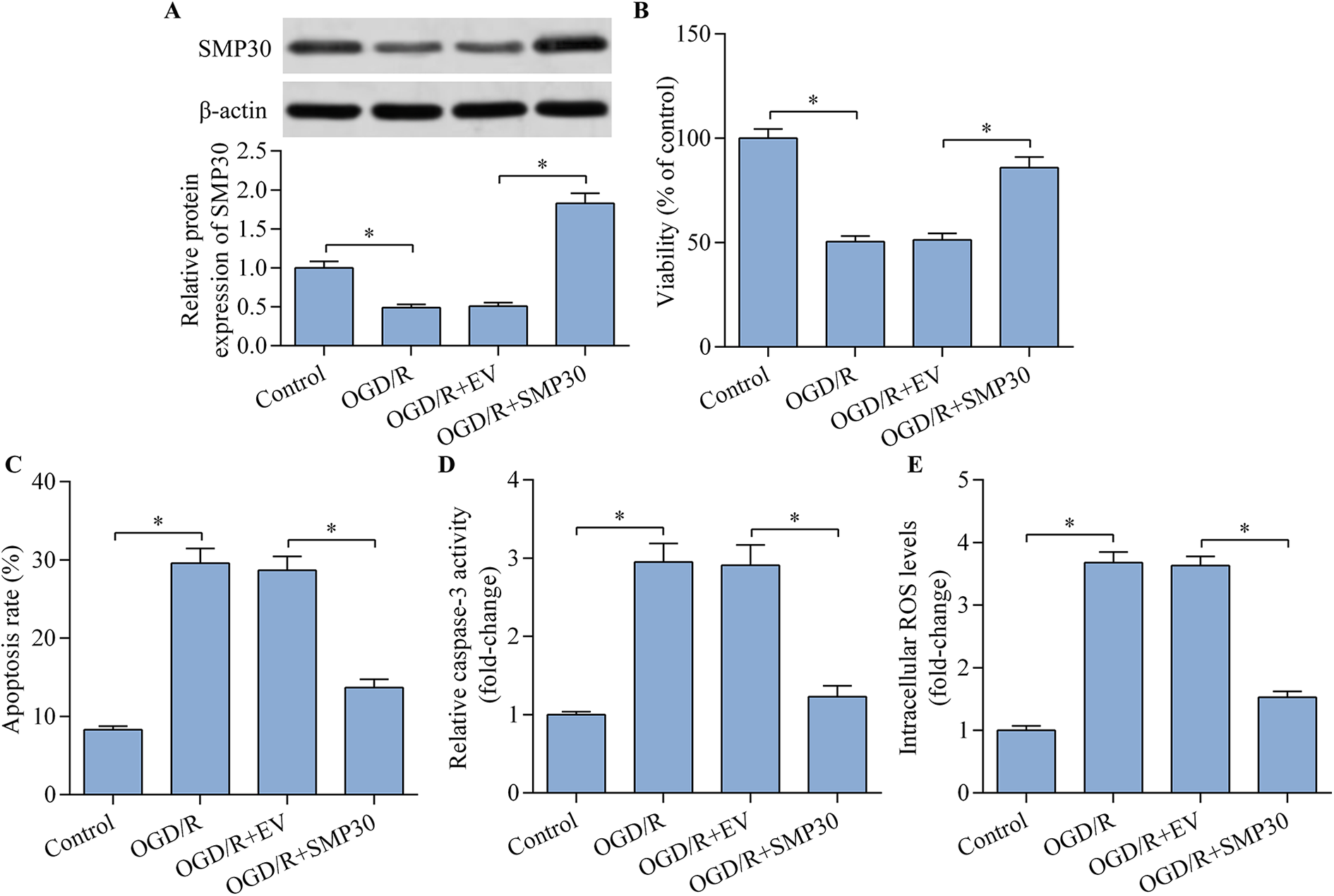
SMP30 overexpression alleviated OGD/R-induced apoptosis and oxidative stress in HT22 neurons. HT22 neurons were transfected with empty vector (EV) or SMP30 expression vector for 48 h prior to OGD/R injury. (A) SMP30 protein expression was determined by western blot. (B) The effect of SMP30 on cell viability was assessed with the CCK-8 viability assay. The effect of SMP30 overexpression on cell apoptosis was measured by (C) Annexin V-FITC/PI flow cytometric analysis and (D) caspase-3 activity assay. (E) The effect of SMP30 overexpression on the intracellular ROS level was detected by flow cytometric analysis. *p < 0.05.
SMP30 modulated Keap1/Nrf2 signaling
A previous study demonstrated that the cytoprotective effect of SMP30 is associated with its regulatory effect on Keap1/Nrf2 signaling. 29 Thus, we determined whether SMP30 is involved in regulating Nrf2 signaling in OGD/R-exposed neurons. SMP30 overexpression markedly increased Nrf2 nuclear protein expression and decreased Keap1 protein expression (Figure 3A and B). Moreover, SMP30 overexpression significantly increased the transcription activity of Nrf2/ARE (Figure 3C). Nrf2/ARE signaling activation by SMP30 overexpression was further confirmed by RT-qPCR analysis which demonstrated that the expression of heme oxygenase-1 (HO-1) and NAD(P)H: quinone oxidoreductase 1 (NQO1), the downstream target genes of Nrf2/ARE, was significantly upregulated by SMP30 overexpression (Figure 3D and E). These results confirm that SMP30 overexpression contributes to Nrf2/ARE activation in OGD/R-exposed neurons.

SMP30 modulated Keap1/Nrf2 signaling. HT22 neurons were transfected with empty vector (EV) or SMP30 expression vector for 48 h prior to OGD/R injury. Protein expression of (A) nu-Nrf2 and (B) Keap1 was determined by western blot. (C) Nrf2/ARE transcriptional activity was monitored with an ARE-dependent luciferase reporter assay. Relative mRNA expression of (D) HO-1 and (E) NQO1 were examined by RT-qPCR. *p < 0.05.
Keap1 overexpression abolished SMP30-mediated activation of Nrf2/ARE signaling
To confirm whether SMP30 contributes to Nrf2 signaling activation via Keap1 downregulation, we detected the effect of Keap1 overexpression on SMP30-mediated Nrf2/ARE activation. Keap1 overexpression significantly reversed the increased Nrf2 nuclear protein expression and Nrf2/ARE activation mediated by SMP30 overexpression (Figure 4A–C). Moreover, SMP30 overexpression-mediated neuroprotection on OGD/R-induced injury was partially reversed by Keap1 overexpression (Figure 4D–F). These results confirm that SMP30 promoted Nrf2 activation via Keap1 downregulation.

Keap1 overexpression abrogated SMP30-mediated activation of Nrf2/ARE signaling. HT22 neurons were cotransfected with Keap1 and SMP30 expression vectors and incubated for 48 h, followed by OGD/R exposure. Protein expression of (A) Keap1 and (B) nu-Nrf2 was examine by Western blot. (C) Nrf2/ARE transcription activity was monitored with an ARE-dependent luciferase reporter assay. (D) Cell viability was assessed with the CCK-8 viability assay. (E) Cell apoptosis was evaluated by the caspase-3 activity assay. (F) Intracellular ROS level was detected by flow cytometric analysis. *p < 0.05.
Nrf2 inhibition reversed SMP30-mediated neuroprotection
To validate whether Nrf2 contributes to SMP30-mediated neuroprotection, we further detected the effect of Nrf2 knockdown on SMP30-mediated effect in OGD/R-exposed neurons. Nrf2 siRNA transfection significantly decreased Nrf2 expression in SMP30 expression vector-transfected cells (Figure 5A). Notably, Nrf2/ARE activation induced by SMP30 overexpression was significantly blocked by Nrf2 knockdown (Figure 5B). Moreover, SMP30 overexpression-mediated neuroprotection against OGD/R-induced injury was markedly reversed by Nrf2 knockdown (Figure 5C–E). Overall, these results suggest that Nrf2 contributes to the modulation of SMP30-mediated neuroprotection in OGD/R-exposed neurons.
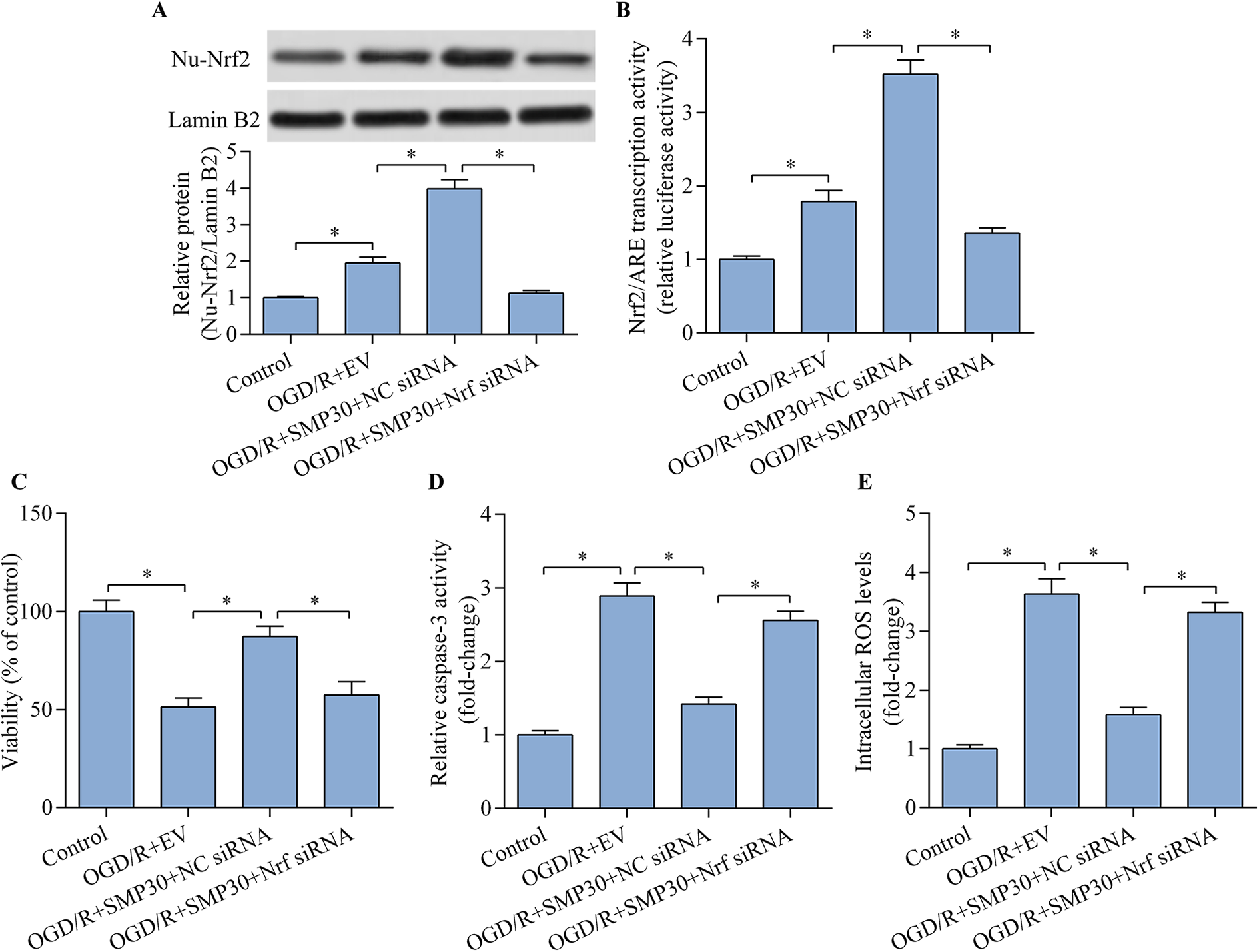
Nrf2 inhibition reversed SMP30-mediated neuroprotection. HT22 neurons were cotransfected with Nrf2 siRNA and SMP30 expression vector for 48 h and then exposed to OGD/R treatment. (A) Nuclear Nrf2 expression was determined by western blot. (B) Nrf2/ARE transcriptional activity was monitored by ARE-dependent luciferase reporter assay. (C) Cell viability was evaluated with the CCK-8 viability assay. (D) Cell apoptosis was detected by the caspase-3 activity assay. (E) Intracellular ROS level was assessed by flow cytometric analysis. *p < 0.05.
Discussion
In the current study, we revealed an important role for SMP30 in regulating OGD/R-induced neuronal injury. Our results demonstrated that SMP30 was decreased in OGD/R-exposed neurons and its overexpression protected these cells from OGD/R-induced apoptosis and oxidative damage. Moreover, our study elucidated that SMP30 overexpression contributed to the activation of Nrf2/ARE antioxidant signaling via downregulation of Keap1, which may be responsible for the SMP30-mediated protective effect (Figure 6). Collectively, our study suggests a neuroprotective role for SMP30 that may serve as a potential target for cerebral ischemia/reperfusion injury.
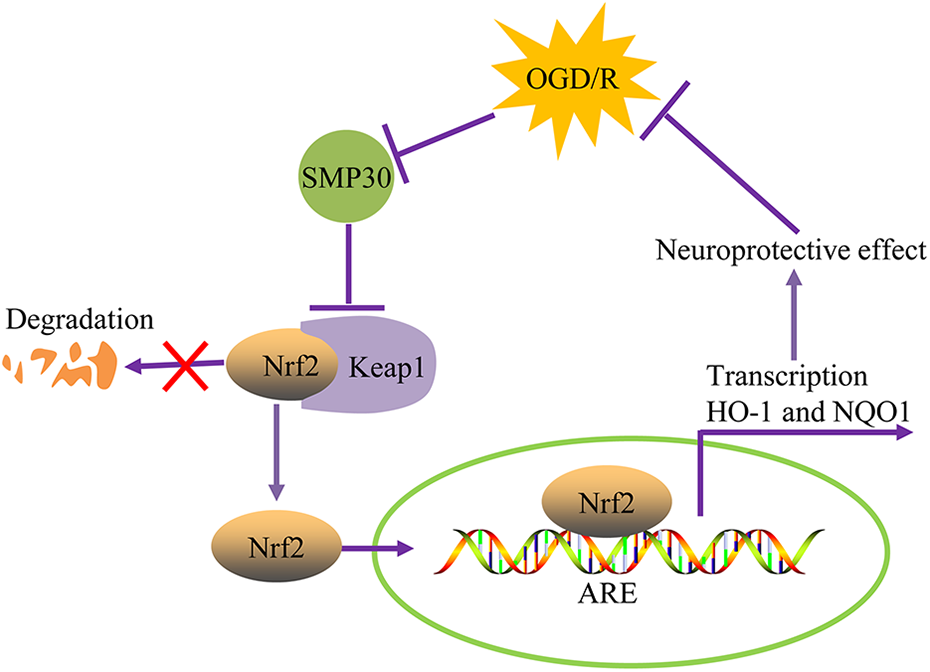
A schematic diagram of SMP30 in regulating OGD/R-induced neuronal injury through Keap1/Nrf2 signaling.
SMP30 downregulation contributes to the pathological processes of various diseases. SMP30 knockout significantly shortens the lifespan of mice. 30 SMP30 knockout mice are susceptible to age-related lung diseases and diabetic nephropathy. 11,31 Cardiac-specific SMP30 overexpression relieves angiotensin-II-induced diastolic dysfunction and cardiac hypertrophy. 32,33 Moreover, SMP30 overexpression protects the heart from doxorubicin-induced cardiac dysfunction. 21 Interestingly, SMP30 knockout in mice exacerbates cardiac injury induced by ischemia/reperfusion. 19 Notably, SMP30 deficiency impairs angiogenesis after limb ischemia. 34 However, little is known about the role of SMP30 in cerebral ischemia/reperfusion injury. In this study, we identified SMP30 as a novel regulator for OGD/R-induced neuronal injury, an in intro model of cerebral ischemia/reperfusion injury. We found that OGD/R injury downregulated SMP30 expression. Moreover, our results demonstrated that SMP30 knockdown exacerbated OGD/R-induced apoptosis and ROS production in HT22 neurons, while SMP30 overexpression alleviated these OGD/R-induced deleterious effects. Therefore, our study suggests a neuroprotective function for SMP30 that may play a potential role in cerebral ischemia/reperfusion injury.
SMP30 dysregulation is associated with the pathogenesis of neurological disorders. SMP30 knockout exacerbates the severity of Parkinson’s disease via impairing astrocyte activation. 35 SMP30 deficiency increases neuroinflammation and anxiety-like behavior in mice. 36,37 Moreover, SMP30 deficiency promotes ROS generation and oxidative stress in brain. 38 Considering that ROS generation is also increased after cerebral ischemia/reperfusion injury, we hypothesized that SMP30 may play an important role in regulating the oxidative stress induced by cerebral ischemia/reperfusion injury. As expected, our results demonstrated that SMP30 overexpression effectively decreased the ROS production induced by OGD/R exposure in HT22 neurons. Our study suggests that SMP30 plays an important role in regulating the oxidative defense in neurons.
A growing body of evidence has reported that SMP30 plays a crucial role in regulating oxidative stress produced under various adverse stimulus. 19 –21 In hydrogen peroxide-exposed cardiomyocytes, SMP30 deficiency augments cardiomyocyte injury by increasing ROS production. 19 SMP30 overexpression represses doxorubicin-induced ROS generation and oxidative DNA damage in cardiomyocytes. 21 In a hepatic steatosis mouse model, SMP30 knockout increases oxidative stress. 20 In this study, our results confirmed that SMP30 was involved in regulating ROS production. Our study, together with previous reports, supports the notion that SMP30 is a key oxidative stress regulator and may serve as a potential target for oxidative stress-associated diseases.
Notably, the cytoprotective function of SMP30 is associated with its regulatory effect on Nrf2/ARE signaling. SMP30 overexpression in colonic epithelial cells increases Nrf2 nuclear translocation by downregulating Keap1-mediated protease degradation of Nrf2. 29 Moreover, activation of Nrf2/ARE signaling by SMP30 protects intestinal epithelial cells from tumor necrosis factor-α-induced apoptosis and inflammatory injury. 29 Consistent with these findings, our results confirmed that SMP30 was a positive regulator of Nrf2/ARE signaling. Our results demonstrated that SMP30 overexpression promoted Nrf2/ARE activation with Keap1 downregulation in HT22 neurons, by which SMP30 alleviated OGD/R-induced apoptosis and oxidative damage. Considering that targeting Nrf2 has become an attractive therapeutic approach for neuroprotection, SMP30-induced Nrf2 activation may have potential application for cerebral ischemia/reperfusion injury treatment.
In conclusion, our results demonstrated that SMP30 overexpression protected from OGD/R-induced neuronal apoptosis and ROS production through modulation of Keap1/Nrf2/ARE signaling, findings that indicate a neuroprotective role for SMP30. Our study highlights the importance of the SMP30/Keap1/Nrf2/ARE axis in regulating OGD/R-induced neuronal injury. These data suggest a potential role for SMP30 in cerebral ischemia/reperfusion injury. Thus, SMP30 could be utilized as a promising target for providing neuroprotection. In the future, it may be possible to develop a feasible approach to manipulate the expression of SMP30 in the ischemic region of brain to evoke Nrf2-mediated neuroprotective effect. However, the exact function and regulatory mechanisms of SMP30 require further validation in vivo using animal models.
Footnotes
Authors' Note
Zhengguo Qiu is now affiliated with Department of Anesthesiology, Affiliated Hospital of Shaanxi University of Traditional Chinese Medicine, Xianyang, Shaanxi, China.
Author contributions
Wenxiong Liu and Haikang Zhao contributed equally to this work and shared the first authorship. Wenxiong Liu and Haikang Zhao: design of the work, performed the experiments and drafted the manuscript. Yuqiang Su, Kefeng Wang, Jing Li, Sha Xue, and Xiaopeng Sun: analyzed the data. Zhengguo Qiu: design of the work, and revised the manuscript. All authors have approved the final version of this article for publication.
Declaration of conflicting interests
The author(s) declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.