
Abstract
Objective
This study aimed to explore the expression and biological functions of SIRT3 in colorectal cancer cells (HCT-116), the impacts of sulforaphane on the ferroptosis of HCT-116 cells and the involvement of the SIRT3/AMPK/mTOR axis in those effects.
Methods
SIRT3-overexpressing (OE) and SIRT3-knockout (KO) cell lines were treated with different concentrations of sulforaphane, RSL-3, and IKE. Cell viability, intracellular ROS, MDA, iron levels, as well as mRNA and protein expressions of target genes were measured.
Results
SIRT3 expression in HCT-116 cells was increased by ferroptosis inducers and decreased by ferroptosis inhibitors. SIRT3 overexpression reduced cell viability and increased intracellular levels of ROS, MDA, and iron, whereas SIRT3 knockdown achieved the opposite effects. SIRT3 overexpression suppressed SLC7A11 expression and promoted the activation of AMPK/mTOR pathway. Restoration of SLC7A11 expression blocked the effects of SIRT3 on ferroptosis induction and cell viability inhibition. SIRT3 effects on cell viability and ferroptosis were antagonized by inhibitors of AMPK or mTOR. Moreover, sulforaphane triggered the ferroptosis of HCT-116 cells by activating the SIRT3/AMPK/mTOR axis.
Conclusions
SIRT3 triggered SLC7A11-mediated ferroptosis in HCT-116 cells, reducing cell viability by activating the AMPK/mTOR pathway, and sulforaphane targets it to inhibit colorectal cancer.
Introduction
Colorectal cancer (CRC) is the third most common cancer type and ranked second for global tumor-associated death, according to the 2020 Globocan database. 1 Metastatic CRC carries a poor prognosis with a median 5-years survival. Immune escape leading to deficient anti-cancer immune response is thought to contribute to CRC progression.2,3 The search for novel therapeutic targets depends on improved approaches to the suppression of CRC cell proliferation.
Ferroptosis describes an iron-dependent form of modulated cell death mediated by excessive lipid peroxidation and has been shown to accompany tumorigenesis and tumor cell proliferation. 4 The involvement of ferroptosis in resistance to chemotherapy drugs has been demonstrated and the triggering of ferroptosis increases chemotherapy sensitivity in CRC. 5 Changes of iron, reactive oxygen species (ROS), and malondialdehyde (MDA) levels in ferroptosis indicate that this mode of cell death is closely associated with cellular metabolism. 6 Solute carrier family 7, member 11 (SLC7A11) participants in the production of GSH, reducing lipid peroxidation and antagonizing ferroptosis. 6 SLC7A11 up-regulation has been reported to be involved in tumorigenesis and metastasis. 7 Indeed, high SLC7A11 expression has been linked to worse prognosis in CRC and SLC7A11 inhibition may enhance the effects of chemotherapy or targeted therapy.8,9 However, the mechanism by which SLC7A11 modulation influences ferroptosis remains poorly understood.
Sirtuin 3 (SIRT3) serves an important role in mitochondrial homeostasis, metabolic regulation, gene transcription, and genome stability. 10 SIRT3 causes AMPK phosphorylation, affecting glucose metabolism and suppressing cancer cell proliferation, 11 giving it an anti-cancer role. However, the mechanism of action of SIRT3 in CRC remains to be demonstrated. The potential for SIRT3 to be a CRC treatment target has been demonstrated by network pharmacology and molecular docking. 12 SIRT3 is also known to modulate the AMPK/mTOR pathway to modulate the progression of non-small cell lung cancer. 13 The current study investigated the impact of SIRT3 on the AMPK/mTOR cascade and ferroptosis in CRC cells. As part of our studies, we explored the mechanism of action of sulforaphane in CRC cells. Derived from cruciferous vegetables, sulforaphane suppresses the survival of various tumors. 14 We examined whether sulforaphane induces the ferroptosis of CRC cells by regulating the SIRT3/AMPK/mTOR axis.
Materials and methods
Cell lines and culture
HCT-116 cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum (EveryGreen, Zhejiang, China) and 1% penicillin/streptomycin (Solarbio, Beijing, China), at 37°C with 5% CO2.
Compounds
RSL-3 (#SML2234), IKE (#329600), ferrostatin-1 (Fer-1, #SML0583), deferoxamine (DFO, #252750), dorsomorphin (DOR, #P5499), rapamycin (RAP, #553210), and sulforaphane (SFN, #S4441) were purchased from Sigma-Aldrich (CA, USA). All compounds were dissolved in DMSO (MP Biomedicals, OH, USA) and stored at −80°C.
Cell transfection
SIRT3-negative control vector (SIRT3-NC), SIRT3-overexpression plasmid (SIRT3-OE), SIRT3-knockout plasmid (SIRT3-KO) and SLC7A11-overexpression vector (SLC7A11) were synthesized by FITGENE CO., Ltd (Guangzhou, China). HCT-116 cells were grown in 12-well plates at a density of 5 × 104 cells/well for 48 h, transfected with 50 ng/mL plasmids using Lipofectamine® 3000 (Invitrogen; Thermo Fisher Scientific, Inc.), according to the manufacturer’s directions, and transfection efficiency assessed by western blotting and qRT-PCR after 48 h.
Cell viability
Cells were cultured overnight in 96-well plates at a density of 4 × 103 cells/well and treated with compounds, as indicated, for 24 h. 10 μL/well Cell Counting Kit-8 (CCK-8) reagent (Engreen, Beijing, China) was added, plates incubated for 2 h and absorbance measured at 450-nm by a microplate reader (BioTek Instruments, Inc.).
Intracellular ROS
Cells were cultured in 12-well plates at a density of 5 × 104 cells/well for 48 h, treated with compounds, as indicated, for 24 h and intracellular ROS concentrations were measured using C11-BODIPY dye (Annoron, Beijing, China). 5 μM C11-BODIPY was added for 30 min, cells harvested, washed twice with PBS and resuspended in 500 μL PBS. Oxidation of the polyunsaturated butadienyl portion of the dye results in a shift of the fluorescence emission peak from 590 nm to 510 nm.
Levels of iron and MDA
HCT-116 cells were cultured and treated with indicated drugs. Cells were harvested into phosphate-buffered saline using a spatula and lysed in 1% Triton (Bio-Tek, Winooski, VT, USA). Cell lysates were centrifuged at 12,000 g at 4°C for 12 min, then the supernatant was assayed for iron and MDA using commercial detection kits (Guangzhou RiboBio, Guangzhou, China) according to the manufacturer’s instructions.
qRT-PCR
Total RNA was extracted by TRIzol® solution (Sangon Biotech Co., Ltd, Shanghai, China) and RNA content and purity analyzed by spectrophotometer (Thermo Fisher Scientifific Inc., MA, USA). DNase I digestion was performed and RNA reverse transcribed into cDNA. PCR was performed using StepOneTM PCR amplifier (Applied Biosystems, USA) with SYBR-green (TAKARA, Japan) in a 10 µl total volume. Reaction conditions were as follows: 95°C for 10 min; followed by 40 cycles of 95°C for 10 s and 60°C for 1 min. Primers were as follows: SIRT3 forward: 5′- GCTACATGCACGGTCTGTCGAA-3′ and reverse: 5′- CAATGTCGGGTTTCACAACGCC-3′; GPX4 forward: 5′- CAGTGAGGCAAGACCGAAGT-3′ and reverse: 5′-GGGGCAG GTCCTTCTCTATC-3′; ACSL4 forward: 5′- ATTGGTCAGGGATATGGGCT-3′ and reverse: 5′-AGAGGAGCTCCAACTCTTCCA-3′; SLC7A11 forward: 5′- TCATTGGAGCAGGAATCTTCA-3′ and reverse: 5′-TTCAGCATAAGACAAAGCTCCA-3′; GAPDH forward: 5′-AGGAGAGTGTTTCCTCGTCC-3′ and reverse: 5′-TGCCGTGAGTGGAGTCATAC-3′. Relative gene expression levels were normalized to GAPDH.
Western blotting assay
Cell were rinsed three times with PBS, lysed with RIPA buffer (Beyotin, Beijing, China) on ice for 30 min, centrifuged and protein concentration measured by BCA protein assay. 50 μg denatured protein was loaded onto 10 or 12 % SDS-PAGE gels and separated proteins transferred to PVDF membranes (Merck Millipore, USA) for blocking with 5% non-fat milk for 1 h at room temperature. Membranes were incubated overnight at 4°C with primary antibodies against SIRT3 (ab217319, Abcam), GPX4 (ab125066, Abcam), ACSL4 (SAB2701949, Sigma-Aldrich), SLC7A11 (ab37601, Abcam), AMPK (ab32047, Abcam), p-AMPK (ab133448, Abcam), mTOR (ab134903, Abcam), p-mTOR (ab109268, Abcam) and β-actin (ab7817, Abcam) and with HRP-conjugated secondary antibody at room temperature for 1.5 h. Signals were visualized by ECL solution (MilliporeSigma).
Statistical analysis
All experiments were performed in triplicate and GraphPad Prism software was used for all analyses. Comparisons between two groups of continuous variables were made by unpaired t test and Wilcoxon test. A value of p < .05 was regarded as significant.
Results
SIRT3 mediated ferroptosis in CRC cells
Expression of SIRT3 mRNA and protein were increased in HCT-116 cells treated with the ferroptosis inducers, RSL-3 and IKE, relative to control, untreated cells (Figure 1(a)–(c)). The ferroptosis inhibitors, DFO and Fer-1, blocked these effects. Simultaneous treatment of cells with ferroptosis inducers and inhibitors resulted in no change to SIRT3 mRNA and protein levels relative to controls (Figure 1(a)–(c)). Ferroptosis appears to induce the upregulation of SIRT3 expression and SIRT3 may be involved in ferroptosis progression. SIRT3 participates in ferroptosis in CRC cells. Total mRNA and protein were extracted from HCT-116 cells after treatment with 5.0 μM RSL-3, 2.0 μM IKE, 1.5 μM DFO or 2.0 μM Fer-1. (a, b) SIRT3 protein in HCT-116 cells measured by western blotting. (c) SIRT3 mRNA in HCT-116 cells measured by qRT-PCR. (d-e) SIRT3 OE and KO in HCT-116 cells were characterized using qRT-PCR and western blotting. (f-g) Viability of SIRT3 OE and KO HCT-116 cells after exposure to different concentrations of RSL-3 and IKE for 24 h, assessed by CCK-8 assay. (H-J) Intracellular ROS, MDA, and iron levels were measured in SIRT3 OE and KO cells after treatment with 2 μM RSL-3 or IKE for 24 h. *p < .05, compared with SIRT3-NC group; #p < .05, compared with SIRT3-OE group.
SIRT3 mRNA and protein were shown to be appropriately up- or downregulated in SIRT3-overexpressing (OE) and SIRT3-knockout (KO) HCT-116 cells relative to negative control (NC) cells (Figure 1(d) and (e)). Treatment with RSL-3 or IKE reduced the viability of HCT-116 cells overexpressing SIRT3 and levels of intracellular ROS, MDA, and iron were increased (Figure 1(f)–(j)). By contrast, viability was increased in SIRT3 KO cells and levels of intracellular ROS, MDA, and iron were lower than in NC cells (Figure 1(f)–(j)). SIRT3 appears to inhibit cell proliferation and promote ROS, MDA, and iron generation to induce ferroptosis in HCT-116 cells.
Overexpression of SIRT3 suppressed SLC7A11 expression and the AMPK/mTOR cascade
SIRT3 overexpression induced the ferroptosis events in HCT-116 cells evidenced by reducing cell viability, increasing the levels of ROS, MDA, and iron, whereas these effects were blocked by Fer treatment (Figure 1(a)–(d)). Levels of SLC7A11 were reduced in SIRT3 OE cells and increased in SIRT3 KO cell (Figure 2(e) and (f)). Expression of other ferroptosis regulators, ACSL4 and GPX4, were unaffected by changes to SIRT3 expression. Thus, SIRT3 may influence ferroptosis in HCT-116 cells via modulation of the SLC7A11 level. Phosphorylation of AMPK and mTOR was induced in SIRT3 OE and inhibited in SIRT3 KO cells (Figure 2(g)), suggesting that SIRT3 promotes ferroptosis by inducing the activation of the AMPK/mTOR axis. SIRT3 overexpression induced SLC7A11, p-AMPK and p-mTOR expression. (a-d) Cell viability, levels of intracellular ROS, MDA, and iron were measured in SIRT3 OE cells after treatment with 2.0 μM Fer-1 for 24 h. (e-f) GPX4, ACSL4 and SLC7A11 in SIRT3 NC, OE and KO HCT-116 cells, measured by qRT-PCR and Western blotting. (g) Phosphorylation of AMPK and mTOR in SIRT3-NC, -OE and -KO cell lines, measured by Western blotting. *p < .05, compared with SIRT3-NC group; #p < .05, compared with SIRT3-OE group.
High expression of SLC7A11 antagonized the effects of SIRT3 on ferroptosis in HCT-116 cells
Overexpression of SLC7A11 in SIRT3-OE cells resulted in increased cell viability compared with SIRT3-OE cells treated with different concentrations of RSL-3 and IKE (Figure 3(a) and (b)). SLC7A11 upregulation also decreased ROS, MDA, and iron levels in SIRT3-OE cells after treatment with RSL-3 or IKE (Figure 3(c)–(e)). SLC7A11 may block the effects of SIRT3 on the induction of ferroptosis and suppression of cell viability. Restoration of SLC7A11 expression blocked the effects of SIRT3 on ferroptosis and cell viability in HCT-116 cells. (a-b) Cell viability in SLC7A11 overexpressing HCT-116 cells transfected with SIRT3-NC and -OE after RSL-3 and IKE treatment for 24 h. (c-e) Intracellular ROS, MDA, and iron levels in SIRT3-NC and -OE cells lines. *p < .05, compared with SIRT3-NC group; #p < .05, compared with SIRT3-OE group.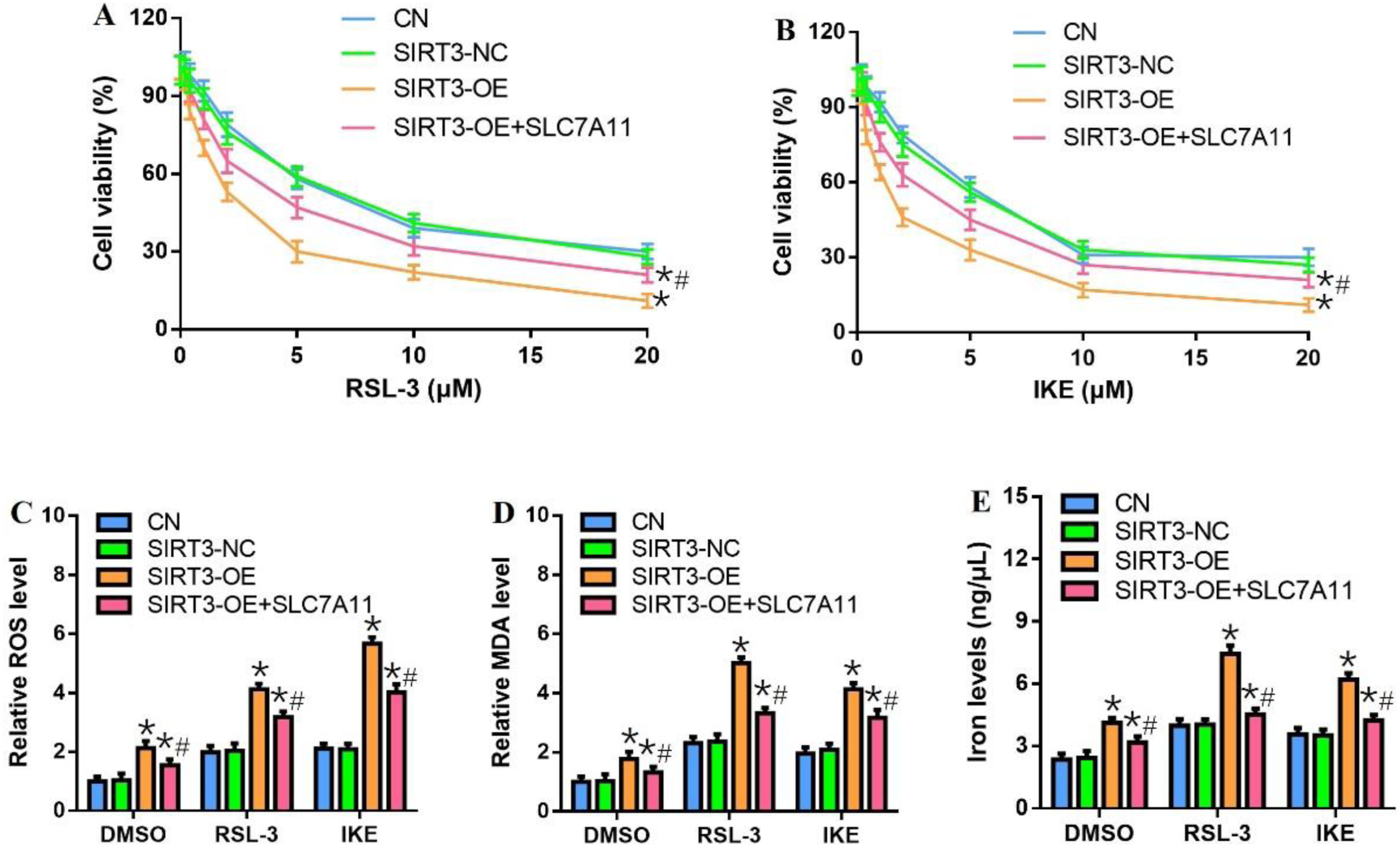
AMPK or mTOR inhibitors antagonized the effects of SIRT3 on SLC7A11 expression and ferroptosis in HCT-116 cells
Overexpression of SIRT3 upregulated p-AMPK, p-mTOR and SLC7A11 (Figure 4(a)), reduced cell viability (Figure 4(b) and (c)) and increased intracellular ROS, MDA, iron levels (Figure 4(d)–(f)). All these effects were antagonized by the AMPK inhibitor, DSP (Figure 4), and by the mTOR inhibitor, RAP (Figure 5). SIRT3 appears to repress SLC7A11 expression and trigger ferroptosis in HCT-116 cells by activating the AMPK/mTOR signaling pathway. DSP antagonized the effects of SIRT3 on SLC7A11 expression and ferroptosis in HCT-116 cells. (a) SIRT3-OE cells were treated with 1.2 μM DSP for 24 h, and AMPK, p-AMPK, p-mTOR, mTOR and SLC7A11 protein measured by Western blotting. (b-c) SIRT3-NC and -OE cell lines were treated with 5 μM RSL-3 and 1.2 μM DSP for 24 h, cell viability were assessed by CCK-8 assay; (d-f) intracellular ROS, MDA, and iron levels were detected. *p < .05, compared with SIRT3-NC group; #p < .05, compared with SIRT3-OE group. RAP antagonized the effects of SIRT3 on SLC7A11 expression and ferroptosis in HCT-116 cells. (A) SIRT3-OE cells were treated with 1.5 μM RAP for 24 h, and p-mTOR, mTOR and SLC7A11 protein measured by Western blotting. (B-C) SIRT3-NC and -OE cell lines were treated with 5 μM RSL-3 and 1.5 μM RAP for 24 h, cell viability were assessed by CCK-8 assay; (D-F) intracellular ROS, MDA, and iron levels were detected. *p < .05, compared with SIRT3-NC group; #p < .05, compared with SIRT3-OE group.

Sulforaphane induced HCT-116 cell ferroptosis by activating SIRT3/AMPK/mTOR signaling pathway
Treating HCT-116 cells with sulforaphane at concentrations from 4 to 16 μM for 12-48 h significantly reduced cell viability at a concentration- and time-dependent way (Figure 6(a)). Moreover, we found that sulforaphane significantly promoted the ferroptosis events by triggering the levels of ROS, MDA, and iron in HCT-116 cells (Figure 6(b)–(d)). It also up-regulated SIRT3, p-AMPK, and p-mTOR expression, and down-regulated SLC7A11 level in a dose-dependent manner (Figure 6(e)). Our data indicate that sulforaphane may induce the ferroptosis of HCT-116 cells by activating SIRT3/AMPK/mTOR signal transduction. Sulforaphane (SFN) induces the ferroptosis of HCT-116 cells and the activation of SIRT3/AMPK/mTOR axis. (a) HCT-116 cells were treated with sulforaphane at different concentrations (0, 1, 2,4, 8, and 16 μM) for 12, 24, and 48 h, cell viability was measured by CCK-8 assay. HCT-116 cells were treated with sulforaphane at different concentrations (0, 4, 8, and 16 μM) for 24 h, (b) ROS, (c) MDA, (d) iron levels, (E) the expressions of SIRT3, p-AMPK, p-mTOR, and SLC7A11 were detected. *p < .05, compared with control group.
To verify that the investigated effects of sulforaphane on the ferroptosis in HCT-116 cells are mediated, at least in part, by SIRT3 and AMPK/mTOR activation, we transfected cells with SIRT3-KO plasmid or treated with AMPK/mTOR inhibitors, then exposed them to 8 μM of sulforaphane for 24 h. SIRT3 knockdown, AMPK inhibitor, and mTOR inhibitor partially reversed the ability of sulforaphane to induce ferroptosis in HCT-116 cells (Figure 7). Our results confirm that sulforaphane induces the ferroptosis of HCT-116 cells by activating SIRT3/AMPK/mTOR axis. SIRT3 knockdown and AMPK/mTOR inhibitors antagonized the effects of sulforaphane on the ferroptosis in HCT-116 cells. Cells transfected with SIRT3-KO plasmid were treated with 8 μM of sulforaphane for 24 h. (a) The activation of AMPK/mTOR, (b) cell viability, (c) ROS, (d) MDA, (e) iron levels were measured. HCT-116 cells were treated with 1.2 μM DSP, 1.5 μM RAP, and 8 μM sulforaphane for 24 h, (f) cell viability, (g) ROS, (h) MDA, (i) iron levels were assessed. *p < .05, compared with control cells; #p < .05, compared with sulforaphane cells.
Discussion
Ferroptosis is an iron-dependent form of cell death, characterized by excessive lipid peroxidation. 10 Dysregulated ferroptosis has previously been shown to be involved in cancer progression.8,9 SIRT3 was found to participate in ferroptosis regulation in the current study. Initiation of ferroptosis resulted in the up-regulation of SIRT3 which promoted the sensitivity of HCT-116 cells to ferroptosis and reduced SLC7A11 expression via activation of the AMPK/mTOR signaling pathway.
SIRT3 has been linked to a variety of pathophysiological processes and is known to be abnormally expressed in many cancer tissues where it participates in tumorigenesis and tumor progression.15,16 Indeed, SIRT3 activated the AMPK/mTOR cascade, an axis controlling tumor cell growth, and reduced proliferation, migration, invasion and angiogenesis in cancer cells in vitro. 17 Low SIRT3 expression has been linked to aggressive features and poor prognosis in patients with non-distant metastatic clear-cell renal cell carcinoma. 18 Thus, SIRT3 is known to influence cell growth and tumorigenesis via several mechanisms but its involvement in ferroptosis has not been previously demonstrated. SIRT3 expression increased after treatment of CRC cells with ferroptosis inducers in the current work and SIRT3 upregulation influenced cell viability, and ROS, DMA, and iron levels in RSL-3 or IKE-treated cells, indicating a role in the modulation of ferroptosis in CRC cells.
GPX4, ACSL4 and SLC7A11 are all known to modulate ferroptosis and inhibition of SLC7A11 expression, in particular, has been shown to trigger ferroptosis in cancer cells and reduce proliferation,19–21 although mechanisms are poorly defined. A previous study has reported that SIRT3-mediated autophagy contributes to ferroptosis-induced anticancer by inducing the formation of BECN1-SLC7A11 complex. 22 Moreover, some researchers have also demonstrated that targeting SIRT3 sensitizes glioblastoma to ferroptosis by promoting mitophagy and inhibiting SLC7A11. 23 SIRT3 overexpression was found to down-regulate SLC7A11 expression and the effects of SIRT3 on ferroptosis and cell viability were reversed in SLC7A11 overexpressing cells during the present study.
The AMPK/mTOR pathway senses intracellular metabolic state, including apoptosis and ferroptosis and an activated AMPK/mTOR cascade has been found in a variety of cancer types, including CRC.24,25 The AMPK/mTOR axis affects tumor progression, including proliferation, metastasis, survival and angiogenesis. 26 SIRT3 deficiency was found to be resistant to autophagy-dependent ferroptosis by inhibiting the AMPK/mTOR pathway. 27 SIRT3 overexpression was found to activate the AMPK/mTOR pathway in the current work and AMPK/mTOR inhibitors antagonized the induction of ferroptosis and inhibition of SLC7A11 expression brought about by SIRT3. We conclude that SIRT3 promoted SLC7A11-mediated ferroptosis in CRC cells by activating the AMPK/mTOR signaling pathway.
Sulforaphane has been shown to suppress the proliferation of tumor cells and induce their apoptosis in several types of cancers, including breast, colon, bladder tumor. 28 Sulforaphane has found to suppress carcinogenesis of CRC via regulating many pathways, such as Wnt/beta-catenin, ERK/Nrf2-UDP.29,30 The present study extends that list to include CRC and ferroptosis. Our study shows that sulforaphane can induce the ferroptosis of HCT-116 cells by regulating cell viability, and we further demonstrate that sulforaphane exerts these effects, at least in part, by activating the SIRT3/AMPK/mTOR pathway.
Conclusion
Our results suggest that SIRT3 modulates CRC cell viability through AMPK/mTOR cascade. Dysregulation of SIRT3 may a novel carcinogenic pathway. Our insights into HCT-116 cell viability, together with our mechanistic experiments with the anti-cancer compound sulforaphane, show SIRT3/AMPK/mTOR to be a potential therapeutic target in CRC.
Footnotes
Acknowledgments
Author contributions
BH and PC conceived and designed the research. LL and WF conducted the experiments. YZ analyzed and interpreted the data. HY drafted the manuscript. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.