
Abstract
Activated Wnt/β-catenin signaling is frequently associated with colorectal cancer. Wnt inhibitors, including tankyrase inhibitors, are being explored as potential anticancer agents. Wnt signaling is also critical for intestinal tissue homeostasis, and Wnt inhibitors have been shown to cause intestinal toxicity in mice by affecting intestinal stem cells. This study sought to characterize the intestinal toxicity of tankyrase inhibitors, including reversibility, and to assess their therapeutic index. Novel tankyrase inhibitor G-631 caused dose-dependent intestinal toxicity with a therapeutic index < 1 after 14 days of dosing in mice. At a tolerated subtherapeutic dose level, the intestinal toxicity was composed of enteritis characterized by villus blunting, epithelial degeneration, and inflammation, which fully reversed after 14 days of recovery. Doubled exposure showed weak antitumor activity in a xenograft colorectal cancer model but also caused more severe intestinal toxicity characterized by multifocal-regionally extensive necrotizing and ulcerative enteritis leading to morbidity or moribundity in some animals. This toxicity was only partially reversed after 14 days of recovery, with evidence of crypt and villus regeneration, mildly blunted villi, and/or scarring in association with chronic inflammation of the submucosa. Therefore, the clinical utility of tankyrase inhibitors is likely limited by the on-target intestinal toxicity and a therapeutic index < 1 in mice.
Introduction
Activation of canonical Wnt signaling is initiated by binding of secreted Wnt glycoproteins to their transmembrane coreceptors, the frizzled proteins (Fz), and the low-density lipoprotein receptor-related proteins 5 and 6 (LRP5/6; Zorn 2001; Figure 1). The Wnt-Fz-LRP5/6 complex transduces a signal into the cell, resulting in nuclear accumulation of a downstream signaling molecule, namely, β-catenin and subsequent activation of T-cell factor (TCF) target genes. Hyperactivation of the Wnt pathway, often in conjunction with mutations in other cell growth regulatory genes, can lead to aberrant cell growth and carcinogenesis (Senda et al. 2007). Approximately 80% of colorectal cancer cells harbor loss-of-function mutations in the tumor suppressor gene adenomatous polyposis coli (APC) and an activated Wnt pathway that leads to increased levels of nuclear β-catenin (Chen et al. 2009). There has been considerable interest in targeting Wnt signaling in cancers. However, pharmacological disruption of β-catenin protein–protein interactions has proven challenging (Polakis 2012). Inhibition of kinases downstream of β-catenin (such as cyclin-dependent protein kinase 8 and casein kinase II) may cause undesired toxicities since these kinases also play a role in non-Wnt pathways. The discovery of tankyrases and their role in stabilizing axis inhibition protein (AXIN) was significant because it may provide new avenues for targeting the Wnt pathway (Polakis 2012). The scaffolding protein, AXIN, is a critical component of the multisubunit destruction complex that tightly regulates the stability of β-catenin. The level of AXIN is regulated by polyadenosine diphosphate (ADP)-ribosylating enzymes tankyrase-1 (TNKS1) and tankyrase-2 (TNKS2), which direct AXIN for ubiquitylation and proteasomal degradation (Chen et al. 2009; Huang et al. 2009; Riffell, Lord, and Ashworth 2012). Inhibition of TNKS1/2 causes AXIN stabilization, leading to enhanced destruction of β-catenin and reduced Wnt signaling (Huang et al. 2009). Several small molecule TNKS1/2 inhibitors have been reported to cause inhibition of Wnt/β-catenin signaling in cell lines as well as antitumor efficacy in selected colorectal cancer human xenograft mouse models (Chen et al. 2009; Huang et al. 2009; Waaler et al. 2011; Waaler et al. 2012; Lau et al. 2013).
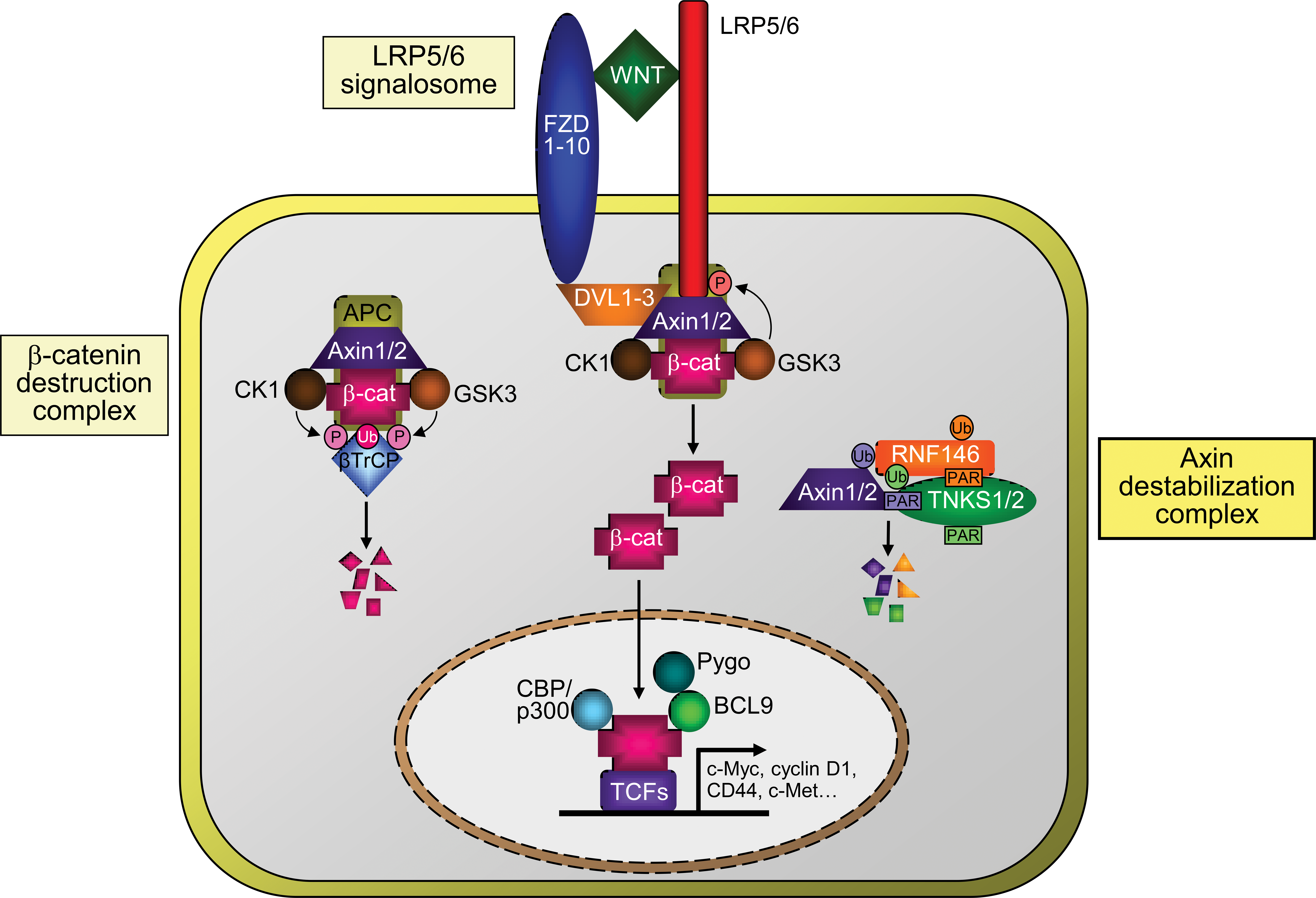
The proposed mechanism by which tankyrase (TNKS) regulates axis inhibition protein (AXIN) stability and Wnt/β-catenin signaling. Activation of Wnt signaling is initiated by binding of secreted Wnt glycoproteins to their transmembrane coreceptors, the frizzled proteins (Fz), and the low-density lipoprotein receptor-related proteins 5 and 6 (LRP5/6). The signal transduction results in nuclear accumulation of β-catenin and subsequent activation of T-cell factor (TCF) target genes. The stability of β-catenin is tightly regulated by the multisubunit destruction complex, where its principal constituents include adenomatous polyposis coli, AXIN, casein kinase 1, and glycogen synthase kinase 3. Under resting conditions, one of the primary functions of this destruction complex is to phosphorylate β-catenin, leading to its ubiquitination and proteasomal degradation. AXIN has been reported to be the concentration-limiting factor in regulating the efficiency of the β-catenin destruction complex. TNKS promotes the ubiquitination and degradation of AXIN by transferring poly(ADP-ribose) to AXIN (poly[ADP-ribosyl]ation). TNKS inhibition has been shown to stabilize AXIN, which in turn leads to β-catenin destruction and attenuated downstream gene transcription (Huang et al. 2009).
Wnt pathway is highly evolutionarily conserved in animals and has been shown to be critical for intestinal tissue homeostasis and self-renewal in rodents, nonrodents, and humans (Pinto et al. 2003; Senda et al. 2007; Hall et al. 2015). Transgenic expression of Dickkopf1 (Dkk1), a potent secreted Wnt antagonist that prevents the formation of the Wnt-Fz-LRP5/6 complex, resulted in the loss of proliferation and villi in the small intestine in mice (Zorn 2001; Pinto et al. 2003). Similarly, transient inhibition of the Wnt pathway using adenoviral expression of Dkk1 in adult mice attenuated proliferation in intestine and colon, causing progressive epithelial degeneration and mortality (Kuhnert et al. 2004). Previously, we reported that severe necrosis and inflammation of the small intestine was seen with a small molecule TNKS1/2 inhibitor G007-LK dosed at high levels in mouse xenograft studies (Lau et al. 2013). The observed reduction in expression levels of proliferation marker (Ki67) and marker for multipotent stem cells of intestinal crypts leucine-rich repeat-containing G-protein-coupled receptor 5 (Lgr5; Barker et al. 2007) are consistent with the notion that the proliferation of intestinal stem cells was perturbed. This intestinal toxicity of G007-LK also raises some important questions regarding the safety of drugs targeting the Wnt pathway. Some of these questions encompass whether the intestinal toxicity is also seen with other tankyrase inhibitors of differing chemotypes, which would suggest that the toxicity is likely on target. If the toxicity is on target, will the proliferation of intestinal stem cells recover following the cessation of treatment with tankyrase inhibitors? Finally, is there a sufficient therapeutic index for tankyrase inhibitor between the activity in colorectal cancer xenograft models and intestinal toxicity?
To address these questions, we dosed mice with a selective small molecule tankyrase inhibitor of a different chemotype. We used a novel TNKS1/2-selective inhibitor G-631 (6-(4-(2,6-difluoro-4-(2-methoxyethoxy)phenyl)piperazin-1-yl)-1-methyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one). To obtain a relevant measure of the therapeutic index, we characterized the efficacy and toxicity of G-631 in a SW403 xenograft colorectal cancer model and its toxicity in naive CD-1 mice. We described in detail the time course and extent of the intestinal toxicity and subsequent recovery. Our data suggest that inhibiting tankyrase causes on-target intestinal toxicity with no safety margin, but the intestinal toxicity appears to be fully reversible at tolerated, subtherapeutic doses. Preliminary account of this work has appeared in the form of an abstract (Zhong et al. 2014).
Materials and Methods
Materials
G-631 (6-(4-(2,6-difluoro-4-(2-methoxyethoxy)phenyl)piperazin-1-yl)-1-methyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one) is a selective TNKS1/2 inhibitor synthesized by Genentech (Feng et al. 2013). For in vitro assays, G-631 was dissolved in dimethyl sulfoxide. For in vivo assays, it was formulated as a spray-dried solid dispersion in hydroxypropyl methylcellulose acetate succinate (HPMC-AS, 18.4% drug load) in corn oil. The control article was HPMC-AS (96.3 mg/ml polymer suspension) in corn oil, with the amount of HPMC-AS being the same as in the high-dose group.
Cell and Biochemical Assays
Tankyrase auto-poly(ADP-ribosyl)ation (PARsylation) biochemical assays were conducted by BPS Bioscience (San Diego, CA) following the BPS PARP Assay kit protocols. Human embryonic kidney 293 (HEK293) cell line was purchased from American Type Culture Collection (Manassas, VA), and cultured according to the supplier’s recommendations. In secondary pharmacology assays, G-631 was tested at 10 μm in the competition binding assay against a panel of 40 targets of receptors, ion channels, amine transporters by CEREP (Poitiers, France). It was also tested in an enzyme assay against acetylcholinesterase (CEREP). In kinase screen, G-631 was tested at 1 μm against 220 kinases by Invitrogen according to their kinase profiling methods (Invitrogen, USA).
Animals
Female nude (nu/nu) mice (6–8 weeks of age; Charles River Laboratories, Hollister, CA) were used in the pharmacokinetic/pharmacodynamic (PK/PD) and efficacy studies. Male CD-1 mice (12 weeks of age; Charles River Laboratories, Raleigh, NC) were used in the toxicity studies. All experiments using mice were approved by the Genentech Institutional Animal Care and Use Committee.
PK/PD and Efficacy Studies in SW403 Xenograft Tumor Model
SW403 cells were cultured in growth media (RPMI 1640, 10% heat-inactivated fetal calf serum, 2 mmol/l
In the PK/PD studies, when tumor volumes reached approximately 400–500 mm3, mice were separated into groups of 4 (control) or 5 (treatment) animals with similar sized tumors. Treatment was initiated a day after grouping the mice, with a single oral dose of either vehicle (5 ml/kg) or G-631 (6.5, 12.5, 25, 50, or 100 mg/kg). Blood samples were taken at 0.5, 1, 2, 8, 16, and 24 hr postoral dosing for PK analysis (n = 4/time point). Blood was collected via retro orbital bleeding procedure under isoflurane anesthesia (interim time points) or via cardiac puncture under CO2 euthanasia (terminal bleed). PK parameters were calculated by noncompartmental methods using WinNonlin version 5.2.1 (Pharsight Corporation, Mountain View, CA).
For efficacy studies, when tumor volumes reached approximately 180 to 250 mm3, mice were separated into groups of 10 animals with similar sized tumors, and treatment was initiated the day after grouping. Oral doses of either vehicle (5 ml/kg/dose) or G-631 (12.5, 25, 50, or 100 mg/kg/dose, once daily [QD]; or 6.25, 12.5, 25, or 50 mg/kg/dose, twice daily [BID]) were administered for 21 days. Tumor volumes were determined using digital calipers (Fred V. Fowler Company, Inc. Newton, MA) using the formula (L × W × W)/2. Group mean ± standard error of mean for tumor volumes and body weights were calculated (n =10). PD analysis of markers including tumor necrosis factor receptor superfamily member 19 (TNFRSF19) expression was performed in tumors collected 2 hr after the last dose. Five mice were randomly selected from the 10 mice in each group for tumor harvesting. Detailed methods on tumor RNA isolation can be found in Lau et al. (2013). Individual data points in quantitative PCR (qPCR) gene expression graph represent relative mRNA levels of TNFRSF19 in tumors of individual mice. Each TNFRSF19 mRNA level was first normalized to the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) level in the same mRNA sample, then normalized to the average of the control vehicle-treated group. Error bars indicated mean ± standard deviation.
Toxicity Study
Three groups of 15 male CD-1 mice were administered an oral dose of 0 (vehicle), 25, or 100 mg/kg/day of G-631 once daily for 14 days. Five mice/group were euthanized at terminal necropsy (after day 14 of dosing), or after a 7- or 14-day recovery period. Hematology and clinical chemistry samples were collected at terminal necropsy (day 15), and after a 7- and 14-day recovery period (day 22 and day 29, respectively) via cardiac puncture under isoflurane anesthesia. Whole blood (approximately 100 µlfor hematology and 600 µl for clinical chemistry) was collected in ethylenediaminetetraacetic acid (EDTA)-containing tubes or serum separator tubes for routine hematology and clinical chemistry evaluations performed on a Sysmex XT-2000iV (Sysmex Corporation, Kobe, Japan) and Beckman AU480 analyzer (Beckman Corporation, Brea, CA), respectively.
For the toxicokinetic analysis, two groups of 12 mice were administered an oral dose of 25 or 100 mg/kg/day of G-631 for either 1 or 7 days. Blood was collected at 1, 4, 6, and 8 hr postdose for toxicokinetic analysis (n = 3/time point).
Toxicity assessments included clinical observations, body weights, clinical pathology, organ weights together with macroscopic and microscopic anatomic pathology. The organ weights were collected for brain, heart, kidneys, liver, lungs, spleen, and thymus. Histopathological examination was performed on bone (sternum), brain, heart, intestines (large and small), kidneys, liver, lungs, spleen, stomach, and thymus. Following euthanasia these tissues were collected, fixed in 10% neutral-buffered formalin, embedded in paraffin, routinely processed as 5-μm thick sections, and stained with H&E.
Plasma G-631 concentrations were determined by liquid chromatography tandem mass spectrometry following protein precipitation with acetonitrile and injection of the supernatant onto the column.
Statistical Analysis
For efficacy and toxicity studies, the treated groups were compared to their corresponding controls using one-way analysis of variance followed by Dunnett’s test as the posthoc test.
Results
In Vitro Assessments and PK Properties of G-631
G-631 is a potent and selective TNKS1/2 inhibitor with favorable in vivo PK properties (Figure 2). In biochemical assays of tankyrase auto-PARsylation activity, G-631 had a biochemical IC50 of 7 nM and cellular potency of 8 nM (HEK293). G-631 is selective against other PARP family members, with IC50 on PARP1 > 10,000 nM. It is also highly selective against kinases and secondary pharmacology targets. G-631 had no >50% inhibition against 220 kinases at 1 μM. On secondary pharmacology panel of 41 targets (receptors, ion channels, transporters, and enzymes), G-631 only had >50% binding inhibition against human norepinephrine transporter (56% inhibition at 10 μm). After a single oral dose, plasma exposure of G 631 reached an area under curve (AUC)0–24 hr of 114 μm × hr and a highest observed plasma concentration of 40.3 μm at 100 mg/kg (Figure 3; Table 1).
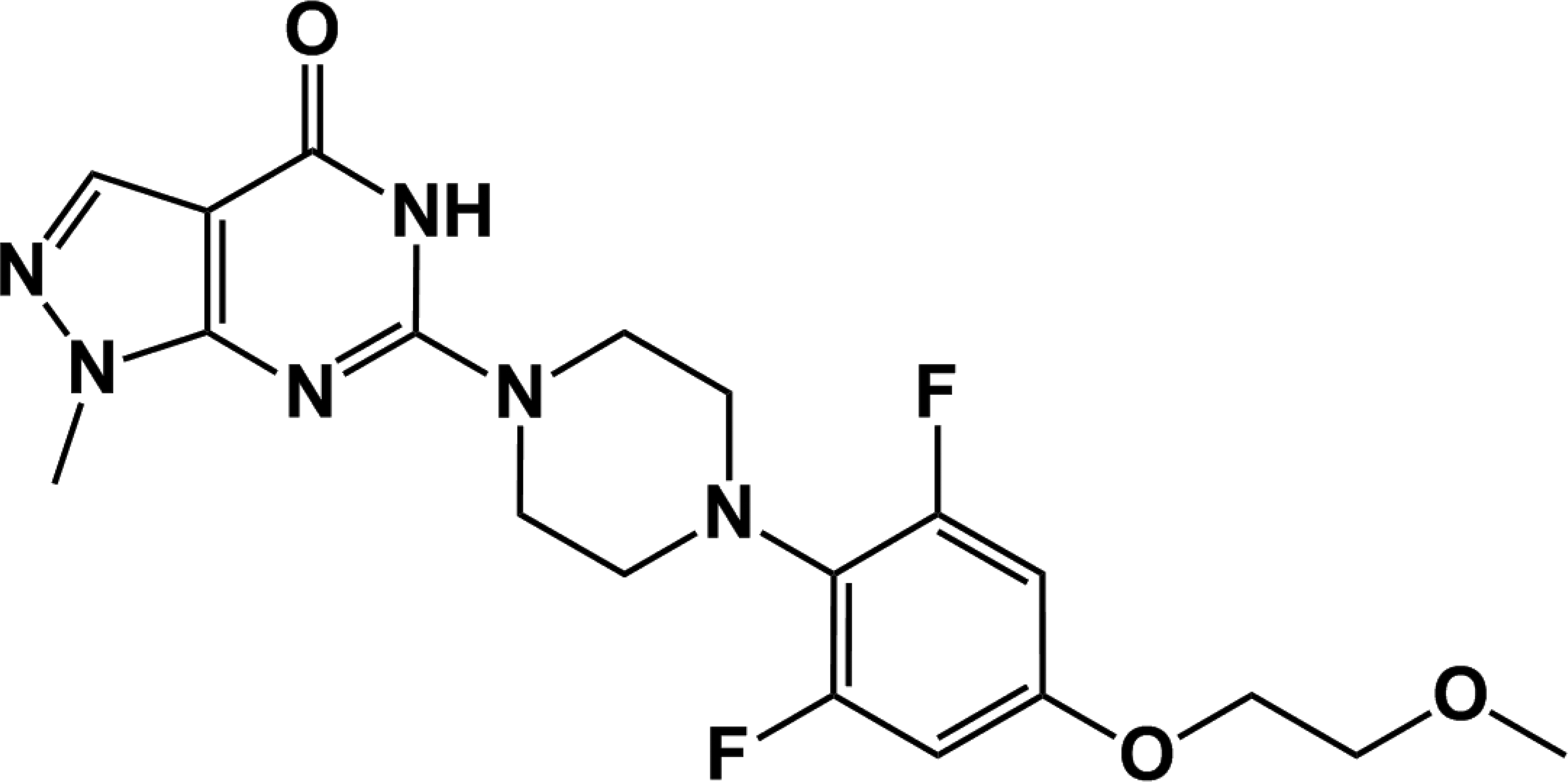
Structure of tankyrase1/2-selective inhibitor G-631.
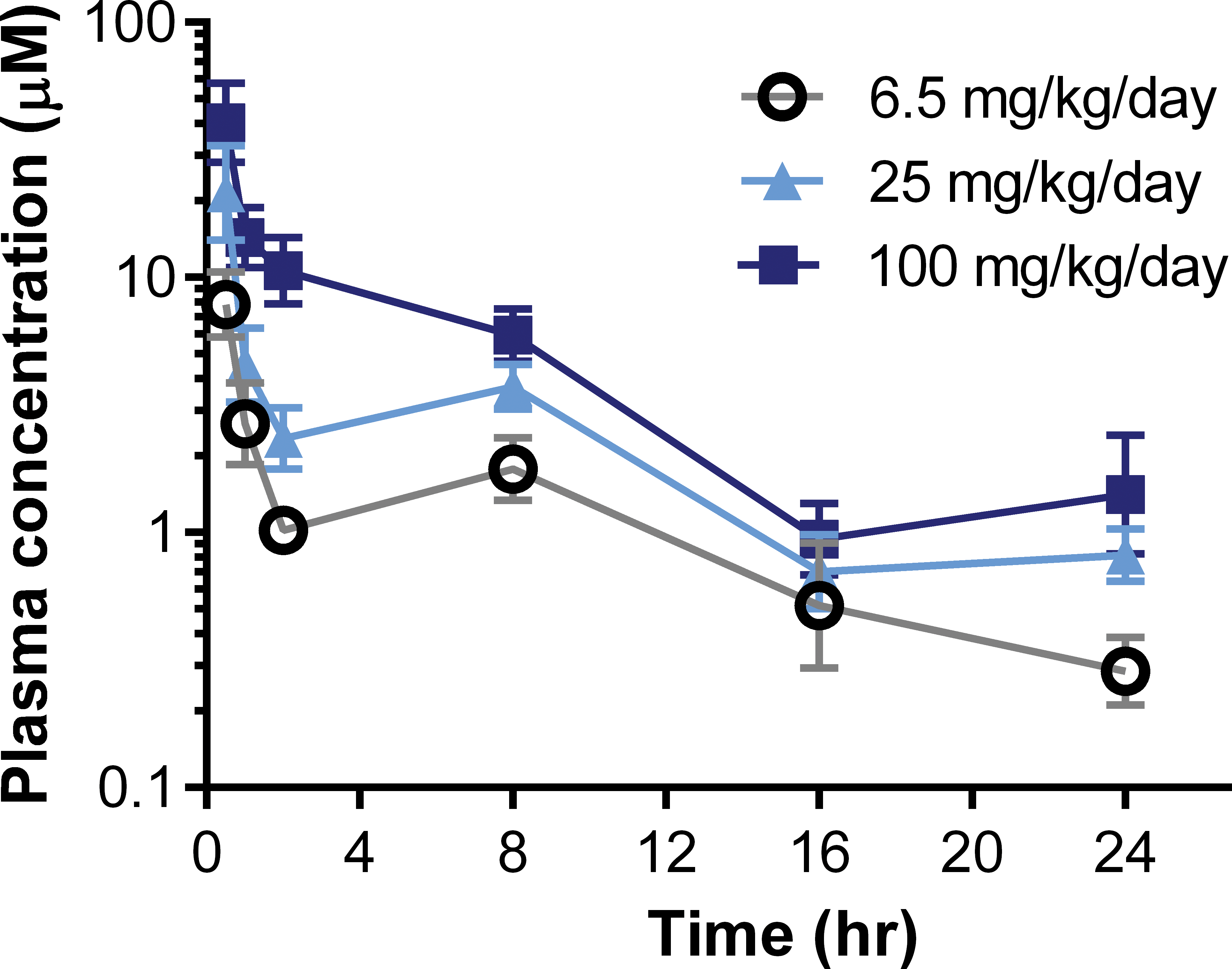
Plasma exposure profile of G-631 following oral administration of a single dose in nu/nu mice implanted with SW403 colorectal adenocarcinoma. The doses given were 6.25, 25, or 100 mg/kg. Data represent mean ± standard error of mean of 4 animals at each time point.
Plasma PK Parameters of G-631 Following Oral Administration of a Single Dose in nu/nu Mice Implanted with SW403 Colorectal Adenocarcinoma.
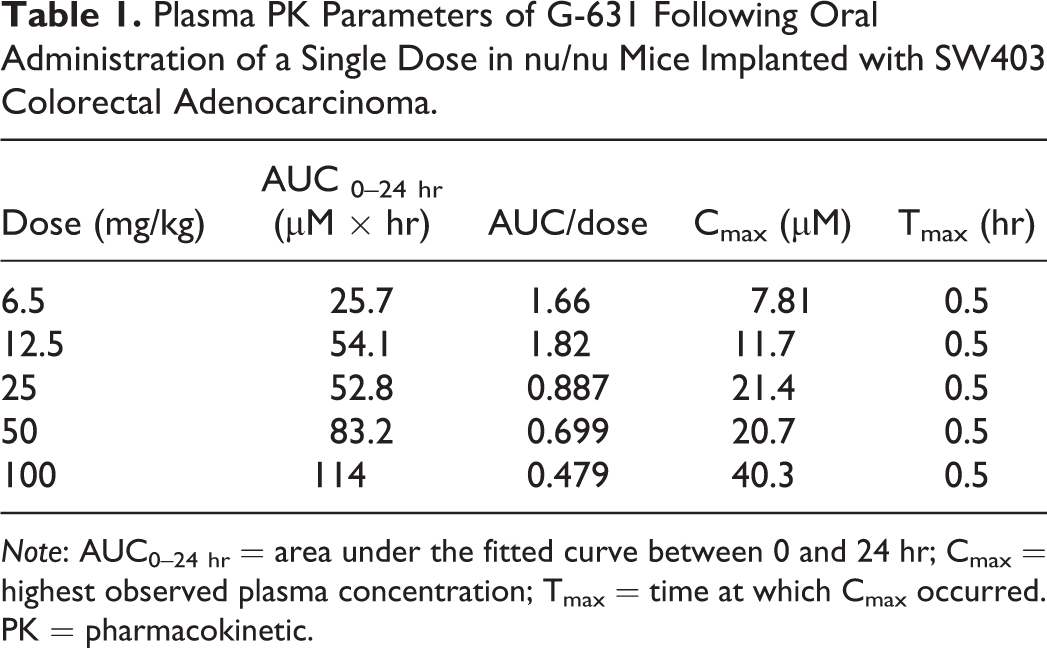
Note: AUC0–24 hr = area under the fitted curve between 0 and 24 hr; Cmax = highest observed plasma concentration; Tmax = time at which Cmax occurred. PK = pharmacokinetic.
Efficacy and Toxicity Assessments
SW403 is a colorectal cancer cell line that harbors the APC mutation. We have previously shown that the SW403 xenograft model was sensitive to the TNKS small molecule inhibitor G007-LK (Lau et al. 2013). Consistent with our previous data with G007-LK, dosing G-631 in the SW403 xenograft model for 21 days resulted in inhibition of Wnt/ β-catenin signaling as indicated by dose-dependent reduction in expression of β-catenin-activated gene TNFRSF19 in the tumors. However, the antitumor activity was weak at the maximum tolerated dose (MTD) of 100 mg/kg/day QD (1,231 mm3 mean tumor volume) versus control (1,604 mm3). There was no overt tolerability issues as indicated by approximately 10% body weight loss at 100 mg/kg/day QD (Figure 4). However, doses of 25 and 50 mg/kg/dose BID were not tolerated, with the observed body weight loss being greater than 10% (data not shown). Gross necropsy of mice presenting with approximately 10% body weight loss showed reddened, edematous, and/or fluid-filled intestinal segments, which was consistent with enteritis. The necropsy results were similar to those observed with G007-LK and were likely the underlying cause of moribundity.
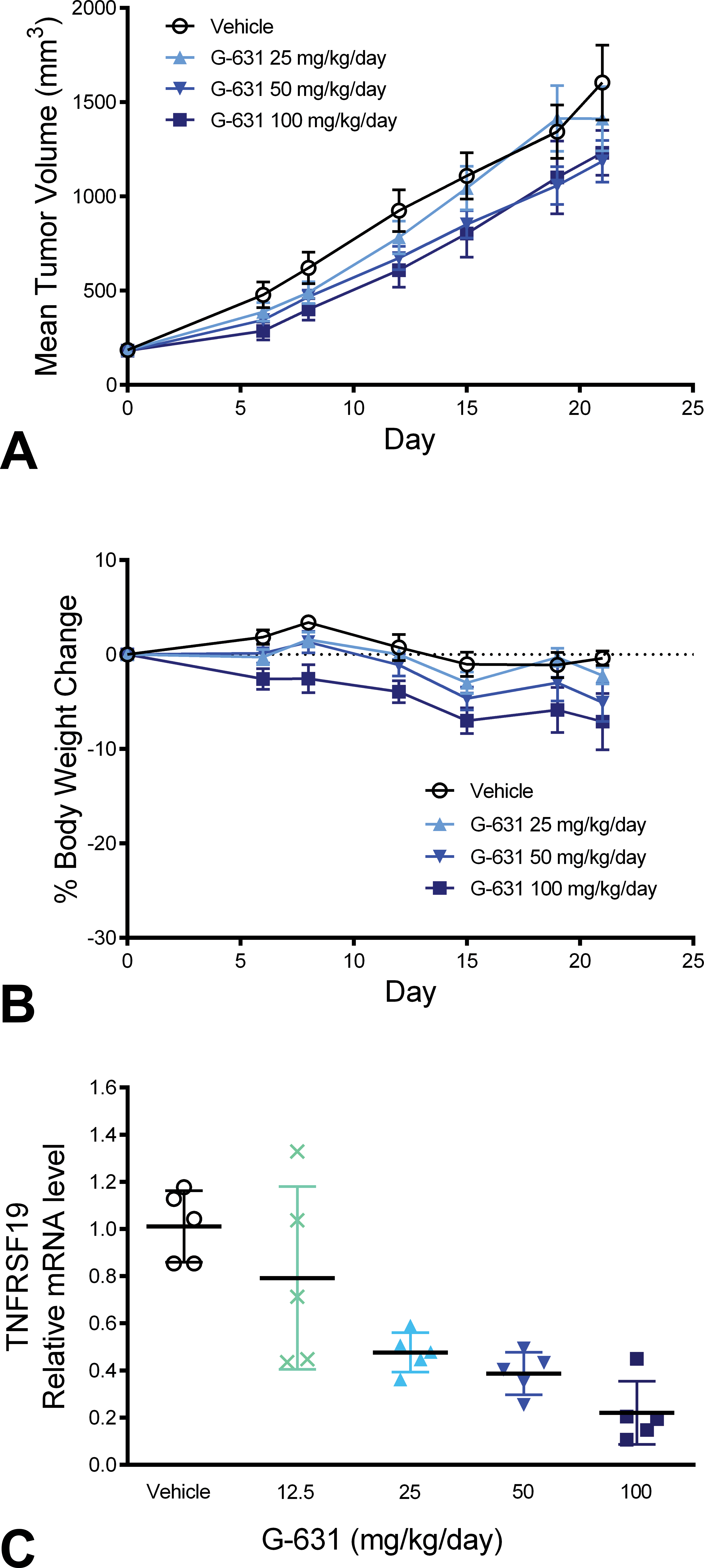
Antitumor efficacy, tolerability, and pharmacodynamics properties of G-631 in SW403 xenograft model. Tumor volume (A), mouse body weight change (B), and relative messenger RNA (mRNA) level of tumor necrosis factor receptor superfamily member 19 (TNFRSF19), a pharmacodynamics marker of the Wnt/β-catenin signaling (C) are shown. G-631 was given once daily orally for 21 days. A and B, each data point represents mean ± standard error of mean of 10 animals. C, each data point represents normalized mRNA level of TNFRSF19 in tumors of individual mouse (n = 5). Group means and standard deviations are also shown.
The intestinal toxicity caused by G-631 at efficacious doses appeared comparable to that of G007-LK (Lau et al. 2013). G007-LK inhibited the proliferation of intestinal stem cells in the crypts with reduction in expression levels of Ki67 and Lgr5, questioning the reversibility of the intestinal toxicity. In order to explore this, we conducted a 14-day repeat-dose study in CD1 mice with two dose levels of G-631. Two dose levels were selected to better understand the dose dependency of the intestinal toxicity. The high dose of 100 mg/kg/day (with weak antitumor activity; Figure 4) was chosen to target the MTD to ensure the presence of intestinal toxicity. The low dose of 25 mg/kg/day (subtherapeutic) was chosen to target approximately half the exposure of 100 mg/kg (Table 1). The epithelial cell turnover rate in murine small intestine is approximately 5–7 days (Marshman, Booth, and Potten 2002), therefore, recovery periods of 7 and 14 days were selected to allow approximately 1 or 2 cycles of cell turnover to occur to be able to detect, repair, and monitor the kinetics of repair.
Plasma exposure from the 14-day toxicity study showed that the animals were continuously exposed to G-631 at both dose levels during the entire treatment period (Table 2). The AUC0–8 hr of 25 mg/kg/day was 56% of that of the 100 mg/kg/day group on day 1. On day 7, the exposure at 25 mg/kg/day was similar to that on day 1 but the exposure at 100 mg/kg/day was lower. These results suggest that there was no accumulation of exposure after multiple days of dosing at 25 mg/kg/day, and the loss of exposure at 100 mg/kg/day by day 7 might be related to the intestinal toxicity which affected compound absorption as described below.
Plasma Toxicokinetic Parameters of G-631 Following Oral Administration at 25 and 100 mg/kg/day QD for 14 Days in Male CD-1 Mice.a
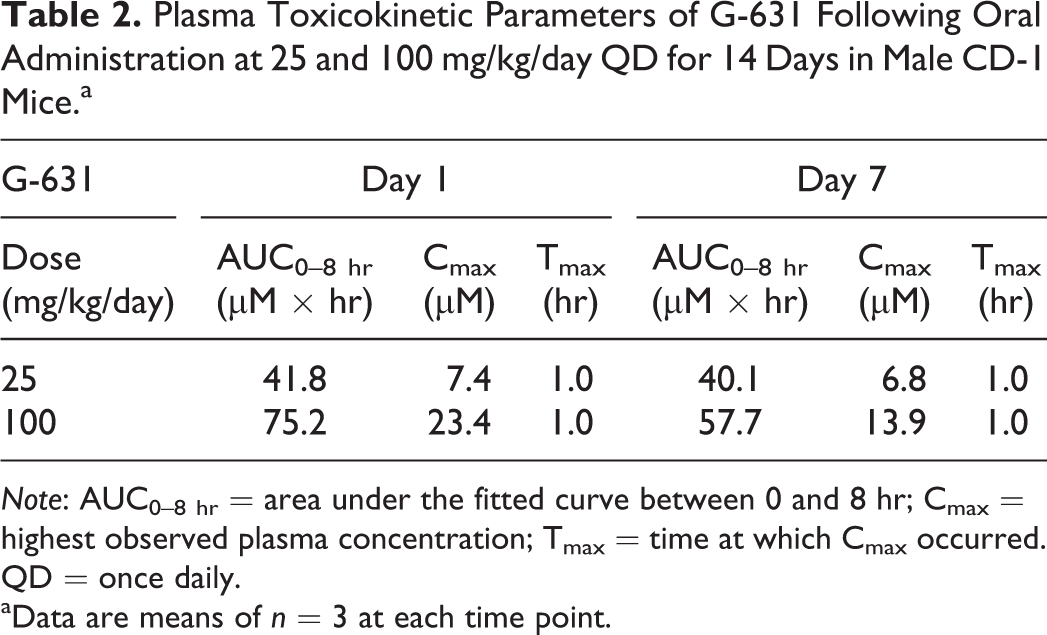
Note: AUC0–8 hr = area under the fitted curve between 0 and 8 hr; Cmax = highest observed plasma concentration; Tmax = time at which Cmax occurred. QD = once daily.
aData are means of n = 3 at each time point.
G-631 dosed at 25 mg/kg/day QD for 14 days was well tolerated in CD1 mice, with no clinical signs, body weight changes, clinical pathology, or gross necropsy findings. Compared with vehicle control, there was minimal-mild small intestine toxicity by the end of dosing (day 15), as shown histopathologically as villus blunting, epithelial degeneration, and mild inflammation in the lamina propria and submucosa (enteritis; Figures 5 and 6). In contrast, G-631 dosed at 100 mg/kg/day QD in CD1 mice was not tolerated, with mortalities observed on days 13, 18, and 20. The cause of death in these mice was attributed to extensive areas of full-thickness mucosal necrosis and/or ulceration and inflammation of the small intestine. Gross necropsy findings at terminal necropsy (day 15) showed reddened, edematous, and/or fluid-filled intestinal segments consistent with enteritis. In general, the ileum was most severely affected, followed by duodenum and jejunum; the cecum and colon are relatively spared. A few lesions appeared in the cecum, but this was mostly edema and may have been related to protein-losing enteropathy rather than primary intestinal toxicity. The histopathology of the intestinal tract after 14 days of dosing was consistent with the gross findings. At 100 mg/kg/day, the findings were moderate marked, characterized by multifocal-regionally extensive areas of mucosal necrosis, ulceration, and marked inflammation involving primarily the small intestines (Figure 6). The large intestine was relatively spared of pathology (not shown). No histopathology changes were noted in the stomach (either squamous or glandular portions; not shown).
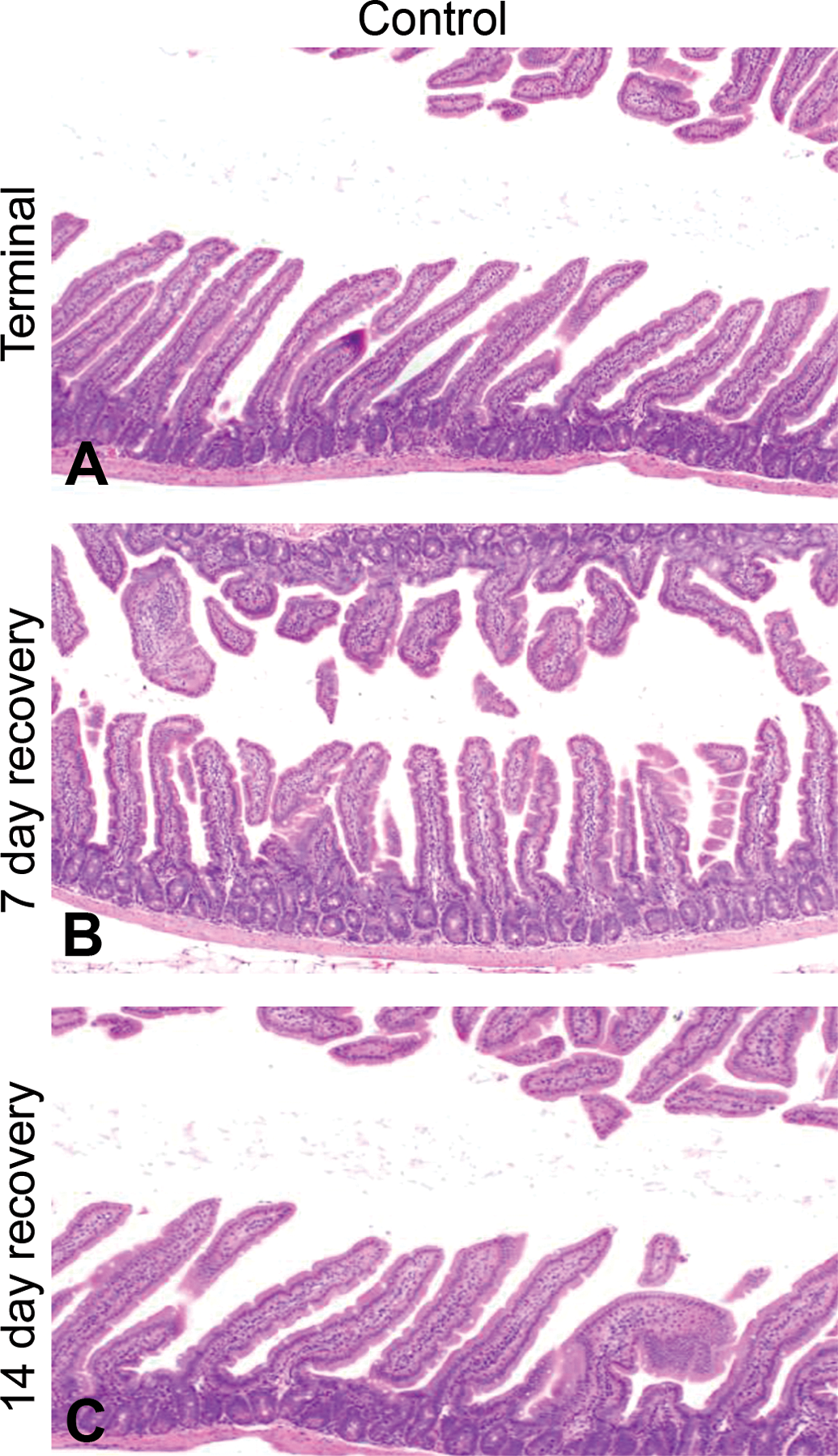
Histopathological characterization of small intestine in vehicle-treated CD-1 mice. Vehicle-treated (control) mouse small intestine section shows normal intestine after (A) 14 days of vehicle dosing, (B) 7 days of recovery, and (C) 14 days of recovery.
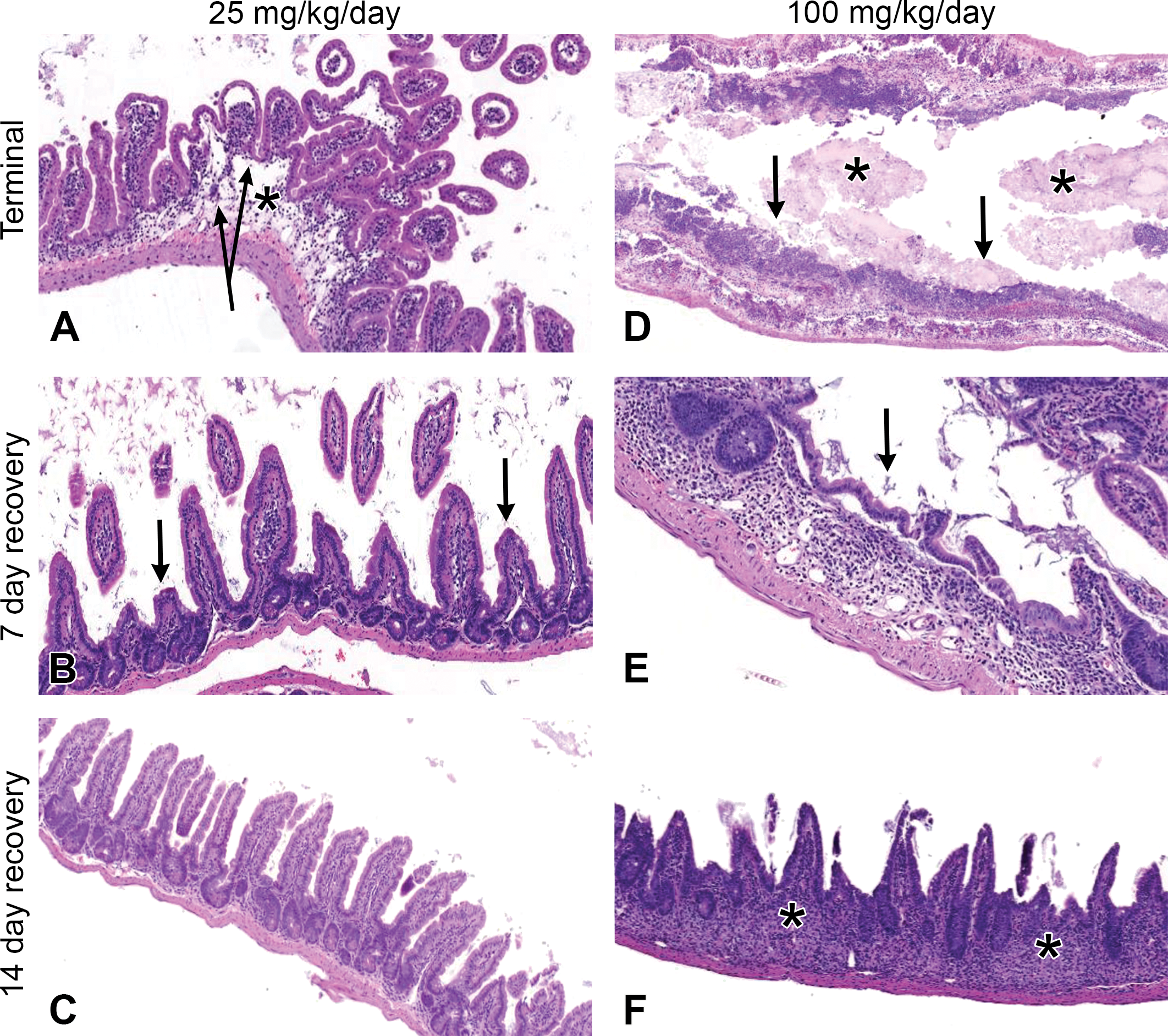
Histopathological characterization of tankyrase inhibitor–related intestinal pathology and reversibility of injury in CD-1 mice. (A) Tankyrase inhibitor–treated mouse small intestine section (25 mg/kg/day, day 14) exhibiting villus epithelial degeneration (arrows), edema, and mixed inflammation of the lamina propria (asterisk). (B) Small intestine after 7-day recovery period showing shortened villi (arrows) indicative of partial recovery from initial intestinal toxicity. (C) Small intestine after 14 days of recovery showing normal villi and full reversibility of the previous intestinal pathology. (D) Tankyrase inhibitor–treated mouse small intestine section (100 mg/kg/day, day 14) exhibiting extensive areas of villus loss, mucosal necrosis/ulceration (arrows), and fibrinonecrotic luminal debris (asterisk). (E) Small intestine after 7-day recovery period showing crypt and villus regeneration (arrow) and/or ongoing repair of ulcers or erosions. (F) Small intestine after 14-day recovery showing shortened villi, submucosal fibroplasia (asterisk), and mixed inflammation; indicating partial reversibility of the intestinal pathology.
The intestinal toxicity of G-631 in mice appeared largely reversible after recovery (Figure 6). Two mice (100 mg/kg/day group) died after 4 to 6 days into the initial recovery period, respectively. The cause of the moribundity is likely due to the diminished intestinal barrier resulting from enteritis potentially causing bacteremia/toxemia as sequelae sustained during the treatment. No moribundity or abnormal clinical signs were present during the latter half of the recovery period. All groups showed moderate gain in body weight during the recovery (Figure 7). There was no macroscopic pathology present in the intestinal tract after a 7- or 14-day recovery.
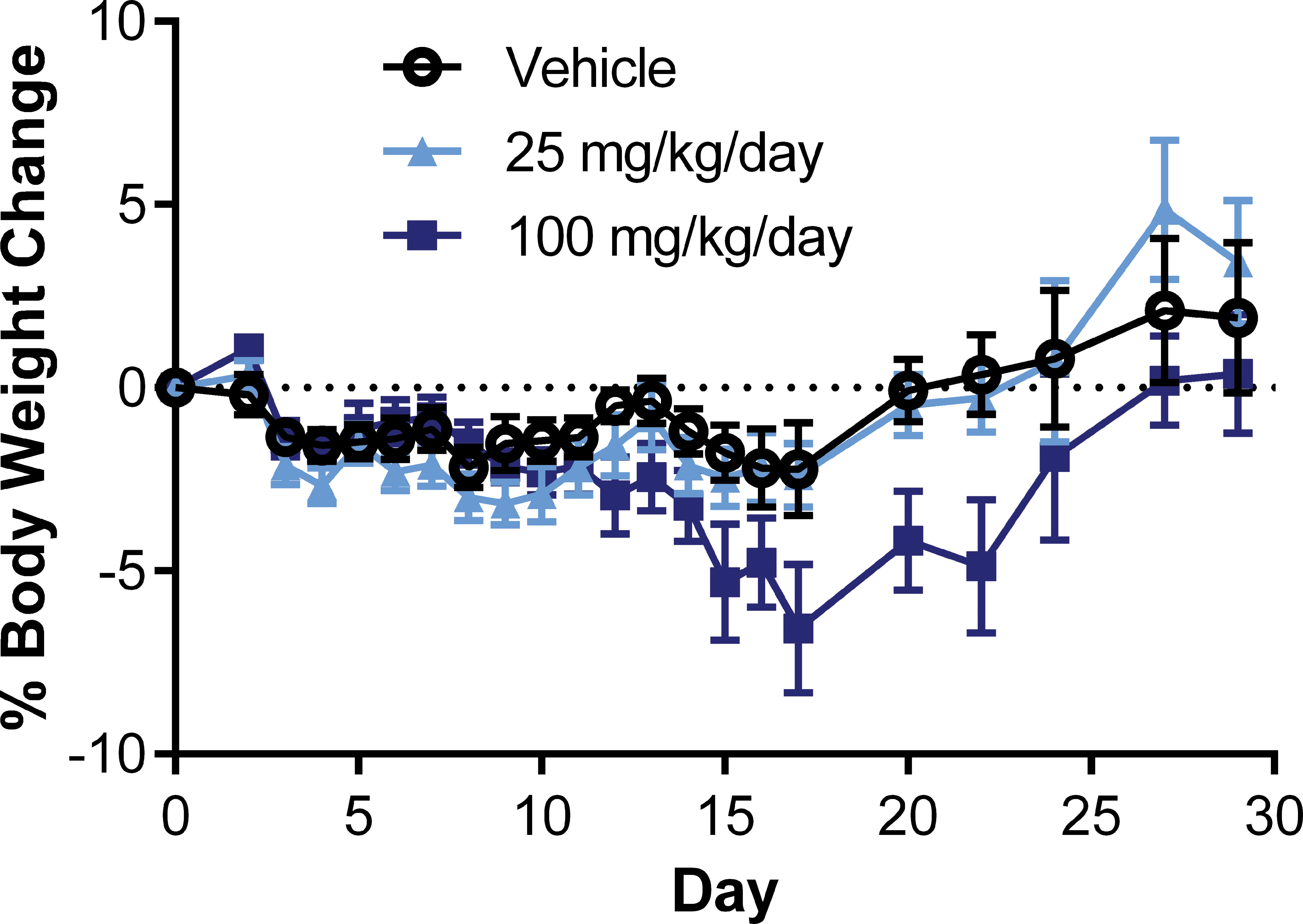
Body weight changes from baseline (day 1) in CD-1 mice administered 25 or 100 mg/kg/day of G-631 once daily for 14 days during treatment and recovery. Each data point represents mean ± standard error of mean for n = 13–15 (days 1–15), n = 6–10 (days 16–22), and n = 3–5 (days 23–29), respectively.
Histopathology findings of the intestinal tract in the 25 mg/kg/day group achieved near complete reversal after 7 days and complete reversal after 14 days (Figure 6). In contrast, the marked intestinal toxicity at 100 mg/kg/day was only partially reversed (Figure 6), with signs of crypt and villus regeneration and/or ongoing repair of ulcers or erosions after 7 days of recovery and mildly shortened villi and/or scarring/inflammation of the submucosa (fibrosis) after 14 days of recovery.
In addition to the intestinal toxicity, G-631 was observed to cause toxicities in other tissues. At 25 mg/kg/day for 15 days, there was myeloid hyperplasia and erythroid hypoplasia of the bone marrow as well as lymphoid depletion in the thymus (data not shown). Treatment of G-631 at 100 mg/kg/day for 15 days caused oval cell hyperplasia and multifocal inflammation in the liver, histiocytic inflammation in the spleen, myeloid hyperplasia and erythroid hypoplasia of the bone marrow, and thymic lymphoid depletion (data not shown). These additional toxicities were not considered the cause of moribundity of these mice at 100 mg/kg and were interpreted as either likely secondary to stress (thymic lymphoid depletion) or intestinal toxicity (liver inflammation).
Clinical pathology findings at the end of dosing (day 15) were limited to the 100 mg/kg/day group (Figure 8). Hematology findings included decreased mean immature reticulocyte fractions (IRF%s) denoting diminished release of early stage erythrocytes from the hematopoietic organs and correlated with the microscopic observations of erythroid hypoplasia in the bone marrow. Absolute neutrophil counts were markedly decreased, monocyte counts were mildly increased, and eosinophil counts were decreased in individual mice. Clinical chemistry findings included markedly decreased total protein and albumin concentrations, alkaline phosphatase (ALP) activities, and moderately decreased globulin concentrations compared to concurrent control values. Cholesterol concentrations were also moderately decreased. There were a few individual instances of minimal increases in absolute red blood cell counts, hematocrit percentage, and hemoglobin concentrations consistent with hemoconcentration due to dehydration. Hematology and clinical chemistry findings at 100 mg/kg/day observed at the end of dosing (day 15) were reversible after 7 days of recovery (clinical chemistry, Figure 8, hematology data not shown).
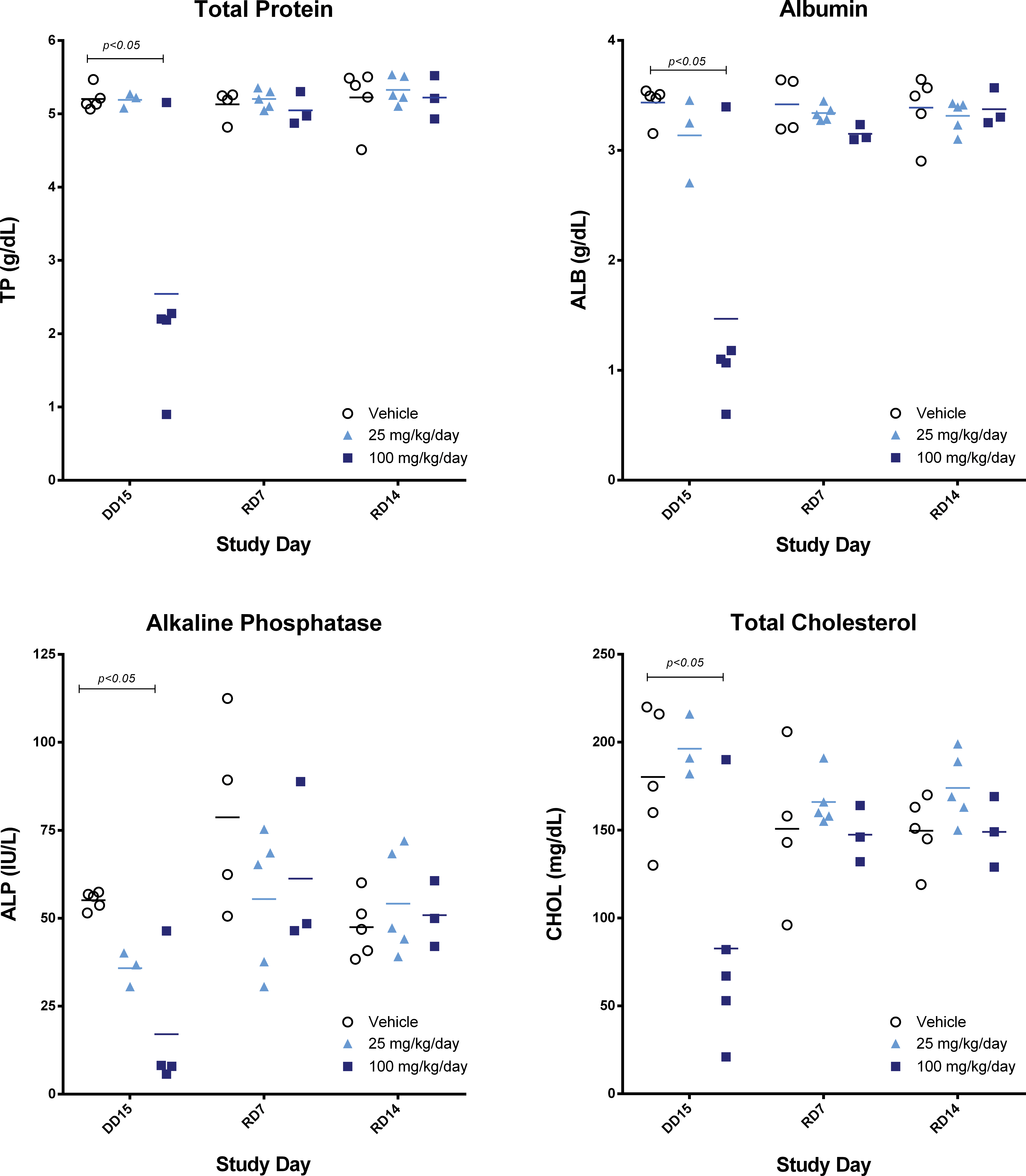
Clinical chemistry parameter changes in mice administered 25 or 100 mg/kg/day of G-631 once daily for 14 days at the end of treatment and recovery periods. Note: DD = dosing day; RD = recovery day.
Discussion
Available literature suggests that hyperactivating the Wnt pathway and accumulating β catenin may contribute to aberrant cell growth and carcinogenesis in colorectal cancer (Senda et al. 2007). Hence, attenuation of this signaling represents a rational and novel targeted approach for the treatment. However, Wnt/β-catenin signaling is also critical for intestinal tissue homeostasis and self-renewal. Inhibiting the pathway with Wnt antagonist Dkk1 or TNKS1/2 inhibitor G007-LK perturbed proliferation of intestinal stem cells and resulted in severe necrosis and inflammation of the small intestine and mortality (Pinto et al. 2003; Kuhnert et al. 2004; Lau et al. 2013).
A successful therapeutic agent requires a sufficient therapeutic index suitable for that indication. The therapeutic index is typically considered to be the ratio of the highest exposure to the drug that results in no toxicity compared to the exposure that produces the desired efficacy (Muller et al. 2012). For life-threatening diseases (typically end-stage oncology indications), a small therapeutic index might be acceptable (Muller and Milton 2012). When evaluating dose-limiting toxicity, it is critical to understand if the toxicity is on or off target and to characterize its dose-dependency, severity, time course, reversibility, and translatability. These data may then be used to inform the clinical development plan including dosing schedule. In this study, we wanted to determine whether the intestinal toxicity caused by the previous tankyrase inhibitor G007-LK was on target or not by testing a novel tankyrase inhibitor G-631 of distinct chemical structure to G007-LK. If similar intestinal toxicity could be reproduced, we wanted to understand whether this toxicity is reversible or not given the fact that proliferation of intestinal stem cell (ISC) is likely perturbed. Furthermore, we wanted to understand whether a sufficient therapeutic index could be achieved, considering the potential target cancer patient population for this molecule. Compounds may cause toxic effects by acting on the primary pharmacological target to elicit on-target toxicity or by acting on known or unknown off-targets to elicit off-target toxicity (Muller and Milton 2012). Since on-target toxicity is driven by the intended target and tightly linked to the occupancy of the primary target, the therapeutic indexes are expected to be small. The off-target toxicities may be related to the chemical, structural, or physicochemical properties such that they are typically considered solvable with chemical modifications. Some of the most direct evidence of a toxicity being on target is the presence of the toxicity with two or more chemically distinct target modulators, ideally from two different chemical series (Stephan-Gueldner et al. 2000; Diaz et al. 2012).
The structure of G-631 is distinct from that of G007-LK, and both were selective with few off targets. But both compounds caused comparable intestinal toxicity. The location and severity of the pathological lesions throughout the intestinal tract were similar. At the subtherapeutic dose of G-631, the intestinal toxicity was minimal to mild. At plasma exposure corresponding to moderate antitumor activity, the intestinal toxicity became moderate to severe, and some animals died within 15 days. Although intestinal concentration of G-631 was higher than that in plasma following oral dose administration, the intestinal toxicity was not likely driven by local concentrations (not shown). This is because similar intestinal toxicity was observed with intraperitoneal administration of G-631 (not shown). The fact that we obtained similar intestinal toxicities and comparable small therapeutic indexes with selective molecules of two distinct chemotypes (G007-LK and G-631) strongly suggests that the intestinal toxicity is on target. This is also consistent with published findings for Wnt antagonist Dkk1 (Pinto et al. 2003; Kuhnert et al. 2004). Taken together, our findings suggest that the intestinal toxicity with tankyrase inhibitors is likely on target, which is consistent with the notion that Wnt/β-catenin signaling is critical for intestinal homeostasis (Hoffman et al. 2004).
In the current 14-day toxicity study, intestinal epithelial degeneration and necrosis with villus blunting and crypt loss were apparent in the small intestine, especially in the ileum at 100 mg/kg/day of G-631 dosing. Peripheral neutrophil counts were markedly decreased in individual mice consistent with recruitment and sequestration at the sites of inflammation and/or decreased production and release by the hematopoietic system. Decreased early stage reticulocytes (IRF%) are a potential on-target effect as studies have demonstrated that Wnt/β-catenin signaling is required for primitive or yolk-sac-derived erythropoiesis (Nostro et al. 2008) and maintenance of the bone marrow microenvironment required to support hematopoiesis (Nemeth et al. 2009). The marked degree of intestinal inflammation and necrosis present may also have contributed to the decreased IRF%s via potential effects on hepcidin regulation (Schmidt 2015). There were markedly and moderately decreased serum albumin and globulin concentrations, respectively, which were considered the result of inflammation and/or decreased absorption (i.e., protein-losing enteropathy). Serum total protein concentrations were subsequently decreased. Cholesterol concentrations were also moderately decreased denoting altered lipid metabolism and/or decreased food consumption. Marked decreases in serum total ALP activities were also present. Unlike in humans where bone and liver isozymes are the main contributors to the total serum ALP activity, mouse total ALP activity is comprised primarily of contributions of the bone and intestinal isozymes (Halling Linder et al. 2013). Intestinal ALP (iALP) is a brush-border-associated protein secreted by enterocytes and its activity is considered a valuable marker of crypt–villus differentiation (Sussman et al. 1989; Fevr et al. 2007). The exact function of iALP is not known, but recently it has been the focus of studies investigating a protective role against lipopolysaccharide-mediated enterocolitis (Heinzerling et al. 2014; Goldberg et al. 2008). We did not have sufficient serum sample volume to differentiate the various alkaline phosphatase isozymes. However, given the known contribution of iALP to total ALP activity in the mouse, the reported loss of function and expression of iALP with starvation/malnutrition (Goldberg et al. 2008), and the observation that with restitution of the intestinal epithelium following the recovery period, alkaline phosphatase activities returned to levels within expected values, the decrease in serum total ALP values at the end of the dosing phase were considered most likely the result of the marked loss of viable enterocytes.
The adult murine intestinal epithelium is characterized by continuous replacement of epithelial cells through a stereotyped cycle of cell division, differentiation, migration, and exfoliation occurring during a 5- to 7-day crypt–villus transit period (Marshman, Booth, and Potten 2002; van Es et al. 2012). Each crypt base contains about 14 long-lived stem cells, which divide symmetrically every day (Snippert et al. 2010; Schepers et al. 2011). This produces, on average, one stem cell (maintaining their numbers) and one daughter cell that enter a population of transit-amplifying cells. The latter migrates upward along the crypt–villus axis while dividing 4 to 5 times (approximately once a day). The transit-amplifying cells exit the mitotic cell cycle and differentiate into one of the five main cell types: mucus-secreting goblet cells, hormone-secreting enteroendocrine cells, antimicrobial peptide, and Wnt-secreting Paneth cells, hydrolase-secreting enterocytes, and opioid-secreting tuft cells. Enterocytes, goblet, enteroendocrine, and tuft cells continue migrating up the villus, whereas Paneth cells migrate downward to reside at the crypt base, where they serve as niche cells to the Lgr5+ stem cells (Marshman, Booth, and Potten 2002). Upon reaching the tip of the small intestinal villi, the cells undergo apoptosis and are exfoliated into the lumen.
Recent publications show that Wnt/β-catenin is a critical growth factor controlling proliferation of crypt stem cells (Kuhnert et al. 2004) as well as differentiation of secretory cells (Pinto et al. 2003). In our previous study (Lau et al. 2013), we described how the tankyrase inhibitor G007-LK caused intestinal toxicity and decreased the proliferation of intestinal stem cells (measured with Ki67 and Lgr5). The data from this study suggest that despite the perturbation of ISC homeostasis, the suppression of epithelial proliferation by tankyrase inhibitors was a reversible process. The extent of reversibility depends on the severity of the initial insult. We found that the mild small intestinal toxicity at 25 mg/kg/day (subtherapeutic) was fully reversed after 14 days of recovery. However, the marked intestinal toxicity at 100 mg/kg/day (beyond MTD) was only partially reversed with evidence of crypt and villus regeneration, mildly blunted villi, and/or scarring/inflammation of the submucosa. It is possible that given adequate recovery period, these intestinal villi may return to normal height and number. However, it is also possible that there could be some permanent submucosal fibrosis secondary to repair of an erosion or ulcer, depending on the severity of the initial injury. Therefore, although the intestinal toxicity at a subtherapeutic and tolerated degree of tankyrase inhibition is expected to be fully reversible after adequate recovery, our findings suggest that tankyrase inhibition at a therapeutic level may cause irreversible intestinal damage. This is consistent with the findings that repressed enterocyte proliferation (measured by Ki67 immunoreactivity) by transient ectopic expression of Wnt antagonist Dkk1 was also reversible (Kuhnert et al. 2004).
The ability of mouse intestines to recover and regenerate following tankyrase inhibitor treatment may be related to the fact that there are two functionally distinct ISC populations with only one sensitive to Wnt modulation (Tian et al. 2011; Yan et al. 2012). Lgr5 marks mitotically active ISCs that exhibit exquisite sensitivity to canonical Wnt modulation and contribute robustly to homeostatic regeneration. In contrast, B lymphoma Mo-MLV insertion region 1 homolog (Bmi1) marks quiescent ISCs that are insensitive to Wnt perturbations and contribute to injury-induced regenerations (Tian et al. 2011; Yan et al. 2012). In experiments where Lgr5-expressing ISCs were ablated by irradiation, the normally quiescent Bmi1+ cells dramatically proliferated, gave rise to Lgr5-expressing cells, and clonally repopulated multiple contiguous crypts and villi, which help to preserve long-term homeostasis of the intestine (Yan et al. 2012). It remains to be seen if similar mechanisms underlie the regeneration of mouse small intestines after treatment with a tankyrase inhibitor.
In contrast to our findings with tankyrase inhibitors and Dkk1, pharmacological inhibition of the Wnt acyltransferase porcupine (PORCN) appeared to be tolerated in a mouse model of Wnt-driven mammary cancer (Proffitt et al. 2013). PORCN is an acyltransferase that adds fatty acid to Wnt, an important step for Wnt activation. In preclinical studies, mice dosed with PORCN inhibitor exhibited no apparent toxicity (including intestines) at therapeutically effective doses (Proffitt et al. 2013). Recently, LGK974, the first small molecule targeting PORCN, entered phase I clinical trials (Lum et al. 2012). More mechanistic studies are needed to understand the difference between Wnt-pathway modulation using PORCN inhibitors (no apparent intestinal toxicity) versus tankyrase inhibitors and Dkk1 (dose-limiting on-target intestinal toxicity).
For oncology indications, the preclinical and clinical therapeutic indexes are often <1, indicating that the toxicity is evident at subtherapeutic exposure (Muller and Milton 2012). Nevertheless, the toxicity needs to be manageable, monitorable, and reversible to protect patient safety. In this study, we observed dose-limiting intestinal toxicity in mice with a selective small molecule tankyrase inhibitor G-631 which is likely on target. While the intestinal toxicity at tolerated doses is fully reversible, these doses also appeared to be subtherapeutic in the mouse model of colorectal cancer, and the severity of the intestinal toxicity precludes determining whether tankyrase inhibition could achieve tumor stasis or regression. Given that intestinal toxicity translates quite well from preclinical species to humans (Olson et al. 2000), and Wnt signaling is evolutionarily conservative, the intestinal toxicity we observed with G-631 and G007-LK in mice may likely translate to humans and limit the therapeutic use of tankyrase inhibitors.
Footnotes
Acknowledgments
The authors thank Mark Zak and Kevin Ford for helpful discussions.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors were employed by Genentech, Inc. when the work was carried out.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.