
Abstract
Objective
Ferroptosis is a newly discovered form of programmed cell death; however, the specific mechanisms that regulate ferroptosis have yet to be fully elucidated in gastric carcinoma. In this study, we aimed to investigate how microsomal glutathione S-transferase 1 (MGST1) regulates ferroptosis in gastric carcinoma cells.
Methods
Gastric adenocarcinoma (SGC7901) cells that overexpressed MGST1 or expressed only low levels of MGST1, were treated with specific compounds (erastin, sorafenib, RSL3, MK-2206 and SC79). Then, we detected the levels of malondialdehyde (MDA), glutathione (GSH), iron and reactive oxygen species (ROS). Protein expression levels of the non-classical autophagy and protein kinase B (Akt)/glycogen synthase kinase-3β (GSK-3β) pathways were determined by western blotting and cell viability was analyzed by Cell Counting Kit-8 (CCK-8) assays. The expressions of target genes were detected using qRT-PCR.
Results
We evaluated a range of ferroptosis-inducing compounds and found that MGST1 expression was down-regulated during ferroptosis in SGC7901 cells. The ferroptosis inducer RSL3 played a role in classical ferroptotic events while the overexpression of MGST1 impaired these effects. Interestingly, the overexpression of MGST1 resulted in the inactivation of autophagy by repressing the expression of ATG16L1 and the conversion of LC3-I to LC3-II. The upregulation of ATG16L1 eliminated the inhibitory action of MGST1 on ferroptosis. Notably, the overexpression of MGST1 induced the activation of the Akt/GSK-3β pathway. An Akt inhibitor antagonized the inhibitory effects of MGST1 on autophagy and ferroptosis.
Conclusion
Collectively, our findings demonstrate a novel molecular mechanism and signaling pathway for ferroptosis. We also characterized that the overexpression of MGST1 induces gastric carcinoma cell proliferation by activating the Akt/GSK-3β signaling pathway.

Keywords
Introduction
Gastric cancer (GC) is one of the most common malignancies and is the leading global cause of death by cancer. 1 Cases of GC are usually at advanced stages when diagnosed; consequently, the opportunity for surgery has already been lost, thus leading to a poor prognosis. 2 Over recent decades, targeted therapies and immunotherapy have become significant new approaches for the treatment of GC. 3 Nevertheless, these strategies are not commonly used for the treatment of GC and the therapeutic effect of these strategies has yet to be fully elucidated. Thus, exploring the molecular mechanisms underlying the occurrence of GC so that we can identify novel therapeutic targets is critical for developing effective new treatments for GC.
Ferroptosis is a genetically encoded program of cell death that involves iron-dependent lipid peroxidation and differs from the classical forms of programmed cell death, such as apoptosis, necroptosis, senescence and pyroptosis.4,5 Extensive mechanistic investigations have demonstrated that autophagy, 6 iron metabolism, 7 ROS metabolism, 8 the mitogen-activated protein kinase (MAPK) signaling pathway and the nuclear factor E2-related factor 2 (Nrf2) signaling are all closely associated with the modulation of ferroptosis.9,10 Moreover, an increasing body of evidence now suggests that ferroptosis is a type of autophagy-associated cell death.11,12 Previous studies have shown that autophagy represents an upstream mechanism that triggers ferroptosis by modulating intracellular iron homeostasis and the production of ROS.13–15 The molecular mechanisms responsible for these events may be associated with several signaling pathways; for example, NCOA4-facilitated ferritinophagy, RAB7A-dependent lipophagy, and HSP90-associated chaperone-mediated autophagy; furthermore, the depletion of Atg5 (autophagy-related 5) has been shown to reduce the intracellular levels of ferrous iron and lipid peroxidation.11–13 In addition, activation of the Akt/GSK-3β signaling pathway has been shown to inhibit autophagy and regulate ferroptosis. 16 Although the mechanisms underlying the modulation of ferroptosis have been widely explored, the specific roles and mechanisms of ferroptosis in the growth of gastric carcinoma cells remain largely understood.
Extensive investigations suggest that the activation of oncogenes, and the inactivation of cancer inhibitor genes both play a role in the progression of GC.17,18 Microsomal glutathione S-transferases-1 (MGST1), a membrane-bound transferase, is mainly localized in the mitochondria, the plasma membrane of the endoplasmic reticulum (ER) and the peroxisomes, and helps to neutralize oxidative stress and protects membranes from lipid peroxidation. 19 MGST1 has also been reported to modulate cell signaling pathways associated with cell growth and apoptosis by modulating the activity of protein kinase B (Akt) and glycogen synthase kinase 3 beta (GSK-3β). 20 A previous analysis of oncogenic patterns and cancer predisposition in GC demonstrated that MGST1 is closely associated with the proliferation of GC cells. 21 Importantly, ferroptosis-associated genes have been demonstrated to predict the prognosis and outcomes of immunotherapy in GC patients, and can also modulate the growth of GC cells. 22 In addition, MGST1 has been reported to repress ferroptotic cancer cell death by binding to ALOX5, thus leading to reduced lipid peroxidation in pancreatic cancer cells. 23 A previous study also showed that the depletion of MGST1 repressed cell growth and induced apoptosis in lung adenocarcinoma cells by inactivating the AKT/GSK-3β pathway. 20 However, the role of MGST1 in GC tumorigenesis and the underlying mechanisms involved remain unclear. Investigating the MGST1-mediated regulation of ferroptosis in GC cells could provide an effective diagnostic factor and therapeutic target for GC. Therefore, in the present study, we investigated novel molecular mechanisms and signaling pathways of ferroptosis in GC cells.
Materials and methods
Cell culture and drug treatment
We acquired a human gastric carcinoma cell line (SGC7901 cells) from the Shanghai Cell Bank, Type Culture Collection Committee of Chinese Academy of Science (Shanghai, China). SGC7901 cells were grown in RPMI 1640 medium (Gibco, NY, USA) supplemented with 10% fetal bovine serum (FBS; Gibco), 100 IU/mL penicillin, and 100 mg/mL streptomycin. Cells were cultured in an incubator with 5% CO2 at 37°C. Erastin, sorafenib, RSL3, MK-2206 and SC79 (Sigma, St. Louis, MO, USA) were dissolved in DMSO and stored in a dark-colored bottle at −20°C. Prior to drug treatment, SGC7901 cells were cultured to 70% confluency and then treated with the indicated drugs for different periods. Cells cultured in RPMI 1640 medium supplemented with an equivalent amount of DMSO but without the test compounds served as a control.
MGST1 and ATG16L1 overexpression
SGC7901 cells were cultured to 90–95% confluency in 6-well plates and then transduced for 48 h with 1.25 μL/well of an expression plasmid encoding MGST1 (pcDNA3.1-MGST1, 1.1 μg/μL; Medicilon Biotechnology, Shanghai, China) or ATG16L1 (pcDNA3.1-ATG16L1, 1.3 μg/μL; Medicilon Biotechnology) together with 1 μL of lipofectamine (Invitrogen, USA). The medium was replaced with fresh medium supplemented with G418 (Medicilon Biotechnology) and stable transfectants were maintained in medium containing 200 μg/mL G418. Cells transduced with the empty pcDNA3.1 vector (Medicilon Biotechnology) served as a negative control group. Cells transduced with the MGST1 and ATG16L1 plasmids considered as experimental group. 24
MGST1 depletion assay
The shMGST1-1 (5′-ACTGCAACTGCATTCTATA-3′), shMGST1-2 (5′-GAAGACTGTGTAGCATTTG-3′) and scramble control (5′-TTCTCCG AACGTGTCACGT-3′) sequences were inserted into the GV248 lentiviral vector, synthesized by Medicilon Biotechnology. The transfection was performed according to a reported protocol. 25 In brief, cells were infected with lentivirus supplemented with polybrene (5 μg/mL) at a multiplicity of infection (MOI) of 20. Cells were collected 96 h after infection for subsequent measurements.
Determination of cell proliferation
For the CCK-8 assays, SGC7901 cells were plated in 96-well plates at a density of 3 × 103 cells/well. When cells were adherent and showed morphological spread, they were exposed to the indicated compounds for 24 h. CCK-8 solution (10 μL) was then added to each well followed by culture for 2 h. Finally, the absorbance at 450 nm was determined with a microplate reader (BioTek Instruments, Winooski, VT, USA). 26
Measurement of MDA levels
MDA levels were detected by a Lipid Peroxidation MDA Assay Kit (Abcam Technology, Cambridge, UK) in accordance with the manufacturer’s directions. Samples and standards were prepared, and the OD values were determined at 532 nm with a microplate reader (BioTek Instruments).
Detection of iron levels
The relative iron level in cell lysates was detected with an Iron Assay Kit (ab83366, Abcam Technology) in accordance with the manufacturer’s directions. The absorbance at 593 nm was measured using a microplate reader (BioTek Instruments).
Determination of GSH levels
GSH levels were determined using a Glutathione Assay Kit (ab65322, Abcam Technology) in accordance with the manufacturer’s directions. The absorbance at 450 nm was measured with a microplate reader (BioTek Instruments).
Measurement of ROS levels
ROS levels were determined with a DCFDA/H2DCFDA-Cellular ROS Assay Kit (ab113851, Abcam Technology) in accordance with the manufacturer’s directions. Cells were analyzed immediately with a fluorescence plate reader (BioTek Instruments) at an excitation wavelength of 485 nm and an emission wavelength of 535 nm. 27
Western blotting assay
Total protein was extracted from cells with RIPA lysis buffer (Cell Signaling Technology, MA, USA) containing PSMF (0.1 mM) and then centrifuged at 12,000 g for 12 min at 4°C. The concentration of protein in each sample was determined with a BCA Protein Assay Kit (Abcam, ab102536) in accordance with the manufacturer’s instructions. Then, western blotting was performed to detect the expression levels of key proteins. In brief, an equal amount of protein (50 μg) was electrophoretically separated by 10% SDS-PAGE and transferred to a PVDF membrane (EMD Millipore, ISEQ00010, Kenilworth, NJ, USA). Next, 5% non-fat milk was used to block non-specific binding sites at room temperature for 1.5 h. After washing, the membranes were then incubated with primary antibodies overnight at 4°C, including MGST1 (ab131059), ATG3 (ab108251), ATG4A (ab108322), ATG12 (ab109491), ATG5 (ab108327), BECN1 (ab207612), ATG7 (ab52472), ATG9A (ab108338), LC3-I (ab63817), LC3-II (ab192890), p-Akt (ab38449), Akt (ab18785), p-GSK-3β (ab93926), GSK-3β (ab280376), and β-actin (ab6276); all antibodies were purchased from Abcam Technology. The following morning, the membranes were washed three times and then incubated with HRP-conjugated anti-rabbit secondary antibody for 1.5 h. Blots were then visualized with an enhanced chemiluminescence detection kit (BIO-RAD, Hercules, CA, USA) and quantified by densitometry using ImageJ. 28
Measurement of mRNA levels
Total RNA was extracted from cells with TRIzol Reagent (Invitrogen, CA, USA) in accordance with the manufacturer’s instructions. First-strand cDNA was then synthesized with HiScript@ II Q RT SuperMix (Invitrogen, CA, USA). qRT-PCR was carried out with ssoFast EvaGreen Supermix (BIO-RAD, Hercules, CA, USA) in a CFX Connect Real-Time PCR Detection System (Bio-Rad). The primers used in our study are listed below. MGST1, F: 5′-ATGACAGAGTAGAACGTGTACGC-3′, R: 5′-TACAGGAGGCCAATTCCAAGA-3′; ATG16L1, F: 5′-ATTTGATGAGCAGTAAACCTCTG-3′, R: 5′-GGGGCTGAAGCATACTTACG-3′; MAPILC3B, F: 5′-ATGCCCTCGGAGAAGAGCTTCAACGCC-3′, R: 5′-CACCTTCGAGCAAAGAGTTGAAGATG-3′; SQSTM1, F: 5′-GACTGATAGTGACCTGTTCGTTGCAAC-3′, R: 5′-CGTGAAGGATGACATCTTCCGAATCTAC-3′; FTH1, F: 5′-GCAGGATATAAAGAAACCAGA-3′, R: 5′-TCTCAATGAAGTCACATAAGT-3′; β-actin, F: 5′-CCTCGCCTTTGCCGATCC-3′, R: 5′-GGATCTTCATGAGGTAGTCAGTC-3′.
The relative expression levels of mRNAs were normalized to β-actin and fold change was calculated by the 2−ΔΔCt method. 29
Co-immunoprecipitation assay
Co-IP was performed with a Classic Magnetic IP/Co-IP Kit (Invitrogen, CA, USA) in accordance with the manufacturer’s instructions. Podocyte lysates were incubated with Co-IP antibodies against ATG12 and ATG5 at 4°C overnight with IgG as a control. Next, protein A/G magnetic beads (GE Healthcare, Chicago, IL, USA) were added to the immune complex solution and incubated for 2 h at 4°C with gently shaking before rinsing with IP wash buffer. The bound proteins were then dissociated from the beads with low-pH buffer for western blot analysis.
Statistical analysis
Each experiment was carried out in triplicate. Results are shown as mean ± standard deviation (SD). Statistical analysis involved either the student’s t-test (two-group comparison) or one-way analysis of variance (ANOVA; comparison of more than two groups). Statistical analysis was performed with SPSS version 17.0 software (SPSS Inc., Chicago, IL, USA) and p < .05 was regarded as statistically significant.
Results
MGST1 expression was downregulated during ferroptosis in SGC7901 cells
Previous studies have reported that clinical (e.g., sorafenib) and pre-clinical (e.g., erastin or RSL3) drugs can induce ferroptosis in cancer cells.30,31 In addition, lipid peroxidation, glutathione (GSH) depletion, excessive ROS production, and redox-active iron accumulation are four crucial events in ferroptosis.
32
As expected, the levels of MDA, iron, and ROS were significantly increased, whereas GSH level and cell viability were strongly reduced in SGC7901 cells after exposure to sorafenib, erastin and RSL3 (Figure 1(a) to (e)). Interestingly, these effects were antagonized by liproxstatin-1 (a potent inhibitor of ferroptosis) but not ZVAD-FMK (a potent inhibitor of apoptosis) and necrostatin-1 (a potent inhibitor of necroptosis) (Figure 1(a) to (e)). MGST1 expression was reduced during ferroptosis in SGC7901 cells. SGC7901 cells were exposed to erastin (10 μM), sorafenib (10 μM), and RSL3 (2.5 μM), with or without associated inhibitors (liproxstatin-1, 100 nM; ZVAD-FMK, 10 μM; necrostatin-1, 10 μM) for 24 h. The levels of (a) MDA, (b) iron, (c) GSH, and (d) ROS were determined; (e) Cell viability was assayed by CCK-8 assays. 
To investigate whether MGST1 is involved in modulating ferroptosis, we measured the protein and mRNA expression levels of MGST1 during ferroptosis in SGC7901 cells. As shown in Figure 1(f) and (g), sorafenib, erastin and RSL3 remarkably repressed the expression of MGST1 at both protein and mRNA levels. However, autophagy inhibitor (3-methyladenine, 3-MA) up-regulated MGST1 protein expression, but not mRNA in RSL3-treated cells (Supplemental Figure 1). These findings indicated that a reduction in MGST1 expression may play a role in modulating ferroptosis in SGC7901 cells. Moreover, MGST1 expression in other gastric cancer AGS cell line was also reduced after treatment with the ferroptosis inducer (Supplemental Figure 2).
The overexpression of MGST1 conferred resistance to ferroptosis in SGC7901 cells
To explore whether the upregulation of MGST1 was directly involved in the inhibition of ferroptosis, we first assessed the expression of MGST1 when overexpressed by a specific plasmid in RSL3-treated cells. As shown in Figure 2, the specific plasmid induced the overexpression of MGST1 while treatment with RSL3 downregulated the expression of MGST1. Indeed, high levels of MGST1 clearly impaired RSL3-induced growth inhibition in SGC7901 cells (Figure 2(b)) with a reduction in a number of ferroptotic events, such as reduced MDA levels (Figure 2(c)), reduced iron levels (Figure 2(d)), GSH production (Figure 2(e)), and ROS elimination (Figure 2(f)). In addition, several inhibitors of ferroptosis (ferrostain-1 or liproxstatin-1) significantly reversed MGST1 knockdown-induced growth inhibition in SGC7901 cells, while ZVAD-FMK, necrosulfonamide (a potent inhibitor of necroptosis), and necrostatin-1, did not strongly antagonize this effect (Figure 2(g)). Collectively, our data suggested that the overexpression of MGST1 conferred resistance to ferroptosis in RSL3-treated SGC7901 cells but not apoptosis and necroptosis. MGST1 overexpression repressed ferroptosis in RSL3-treated SGC7901 cells. SGC7901 cells over-expressing MGST1 were exposed to RSL3 (2.5 μM) for 24 h. (a) The levels of MGST1 protein were measured by western blotting assay. (b) Cell viability was detected by CCK-8 assays. The levels of (c) MDA, (d) iron, (e) GSH, and (f) ROS were assayed. (g) MGST1-deficient SGC7901 cells were exposed to the indicated inhibitors (ferrostatin-1, 1 μM; liproxstatin-1, 100 nM; ZVAD-FMK,10 μM; necrostatin-1, 10 μM; necrosulfonamide, 0.5 μM) and RSL3 for 24 h, and cell growth inhibition was assessed. n = 3 in each group. *p < .05, compared with control group; #p < .05, compared with vector + RSL3; + p < .05, compared with MGST1 group.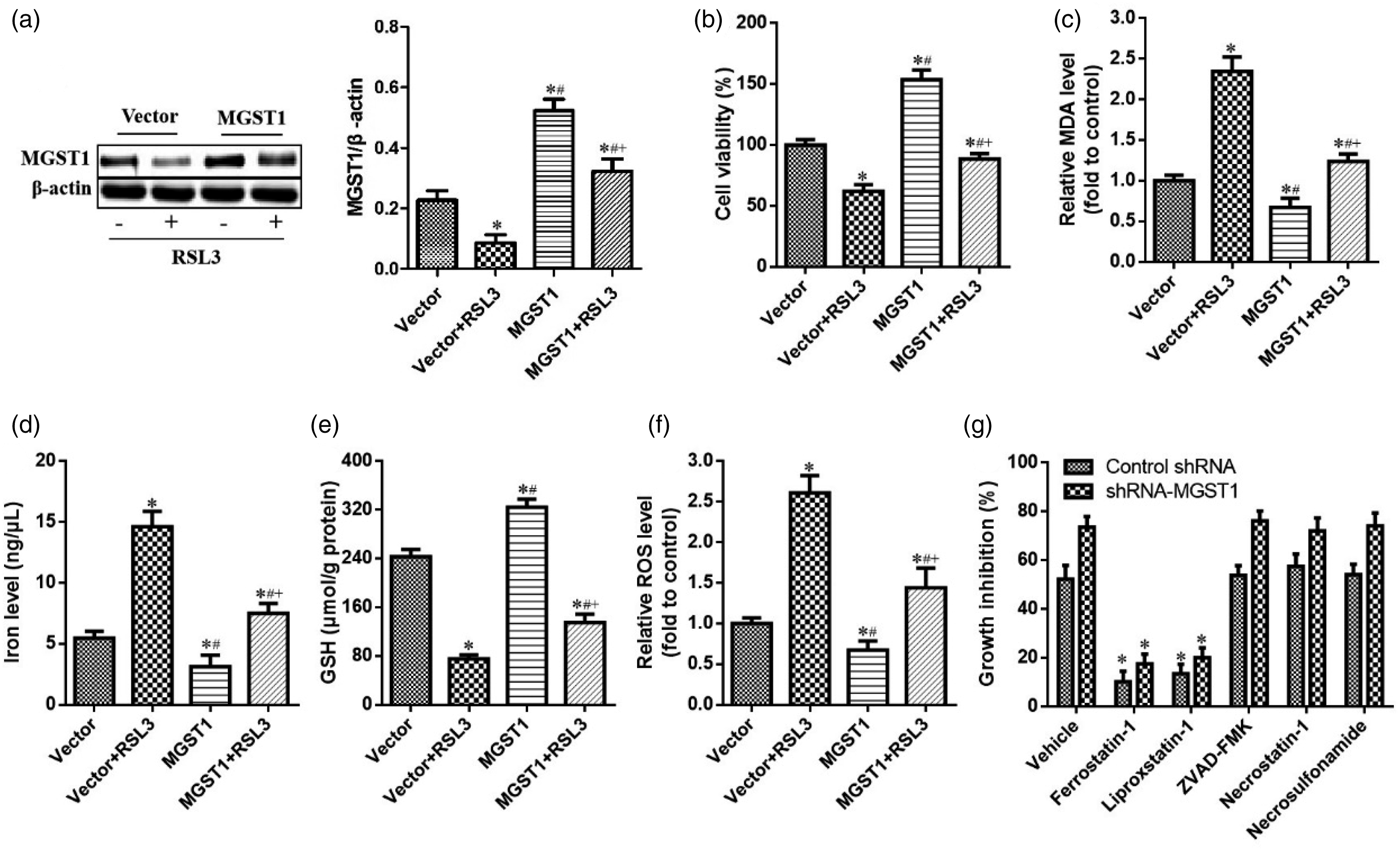
The reduction of ferroptosis induced by the overexpression of MGST1 was correlated with the inactivation of autophagy
To further investigate the molecular mechanisms underlying MGST1-triggered ferroptosis resistance in SGC7901 cells, we detected the expression levels of MGST1 targets involved in the non-classical autophagy pathway.
33
As shown in Figure 3(a), the overexpression of MGST1 significantly inhibited the expression of ATG16L1 but without significant changes in the expression levels of ATG3, ATG4A, ATG12-ATG5, BECN1, ATG7 and ATG9A in SGC7901 cells. In contrast, treatment with RSL3 remarkably induced the expression of ATG16L1 but did not have any significant effects on the expression of other genes related to autophagy (ATG). In this figure, we found the expression of ATG16L1 in MGST1 group was significantly decreased compared with vector group (the fold change was 3.24); the level of ATG16L1 in MGST1 + RSL3 group was lower than vector + RSL3 group (the fold change was 1.57). MGST1 overexpression repressed ferroptosis in SGC7901 cells by modulating the autophagy signal pathway. SGC7901 cells overexpressing MGST1 were exposed to RSL3 (2.5 μM) for 24 h. (a) The levels of proteins related to autophagy (ATG) were then detected by western blotting. (b and c) Formation of the ATG12–ATG5-ATG16L1 protein complex was measured by immune-coprecipitation. (d) The protein levels of LC3-I/II were analyzed by western blotting. n = 3 in each group. *p < .05, compared with vector group; #p < .05, compared with vector + RSL3; + p < .05, compared with MGST1 group.
It is worth noting that the activity of the conserved ATG12-ATG5-ATG16L1 complex is very important for the conversion of LC3-II and the formation of autophagosomes. 34 As expected, immunoprecipitation experiments suggested that the overexpression of MGST1 significantly repressed formation of the ATG12-ATG5-ATG16L1 protein complex, while RSL3 treatment antagonized this effect in SGC7901 cells (Figure 3(b) and (c)). Moreover, high expression levels of MGST1 markedly reduced the conversion of LC3-I to LC3-II, whereas RSL3 treatment significantly induced LC3-II conversion in SGC7901 cells (Figure 3(d)). Collectively, our results support the hypothesis that the inhibition of ferroptosis by MGST1 overexpression was related to the inactivation of autophagy.
The upregulation of ATG16L1 antagonized the inhibitory effect of MGST1 on ferroptosis
To further confirm the regulatory effect of ATG16L1 on MGST1 downregulation-mediated ferroptosis, we used the ATG16L1 plasmid to stimulate autophagy activation for reverse validation. Western blotting assays suggested that the ATG16L1 strongly induced the expression of ATG16L1 and the conversion of LC3-I to LC3- II in SGC7901 cells, whereas repressed autophagic flux and reduced the autophagic degradation of ferritin, but partially impaired MGST1 plasmid-triggered ATG16L1 down-regulation (Figure 4(a)). Indeed, MGST1 overexpression greatly stimulated cell viability (Figure 4(b)) and GSH levels (Figure 4(c)), whereas the upregulation of ATG16L1 antagonized these effects. Similarly, the overexpression of MGST1 repressed other several typical ferroptotic events involving MDA and iron; these effects were also weakened by the ATG16L1 plasmid (Figure 4(d) and (e)). Moreover, the overexpression of MGST1 inhibited apoptotic events involving Bax and Bcl-2; these effects were also weakened by the ATG16L1 plasmid (Supplemental Figures 3A-B). Collectively, our data suggested that the induction of autophagy by the ATG16L1 plasmid impaired the resistance of MGST1 to SGC7901 cell ferroptosis. The upregulation of ATG16L1 antagonized the inhibitory effect of MGST1 on ferroptosis. SGC7901 cells were transduced with MGST1 and the ATG16L1 plasmid; (a) then, the protein expression levels of ATG16L1, MAPILC3B, SQSTM1 and FTH1 were measured by western blotting. (b) Cell viability was evaluated with the CCK-8 assays. (c to e) The levels of MDA, iron, and GSH were analyzed. n = 3 in each group. *p < .05, compared with vector group; #p < .05, compared with ATG16L1 group; + p < .05, compared with MGST1 group.
MGST1 promoted the inactivation of autophagy by inducing the Akt/GSK-3β signal pathway
To elucidate the mechanisms underlying the regulatory effect of MGST1 on autophagy inactivation, we next investigated the signal transduction of the Akt/GSK-3β pathway. As shown in Figure 5(a) and (b), the overexpression of MGST1 significantly increased the expression levels of p-Akt and p-GSK-3β while the depletion of MGST1 clearly suppressed the phosphorylation levels of Akt and GSK-3β. To further confirm that MGST1 modulates ferroptosis in SGC7901 cells by activating the Akt/GSK-3β axis, we treated cells that expressed low levels of MGST1 or cells that overexpressed MGST1, with an Akt agonist (SC79, 1.2 μM) and an inhibitor (MK-2206, 2.0 μM), respectively. As shown in Figure 5(c), MGST1 depletion promoted ATG16L1 expression and the conversion of LC3-I to LC3-II, whereas these effects were antagonized by SC79. In contrast, the overexpression of MGST1 repressed ATG16L1 expression and the conversion of LC3-I to LC3-II; these latter effects were antagonized by MK-2206 (Figure 5(d)). Overall, our findings indicated that MGST1 induces the inactivation of autophagy by activating the Akt/GSK-3β signal pathway. MGST1 mediated the inactivation of autophagy by modulating the Akt/GSK-3β axis. (a) The expression levels of MGST1, p-Akt, Akt, p-GSK-3β, and GSK-3β in SGC7901 cells overexpressing MGST1 were analyzed. (b) The expression levels of MGST1, p-Akt, Akt, p-GSK-3β, and GSK-3β in SGC7901 cells with low-expressing MGST1 were detected. (c) ATG16L1 expression and the conversion of LC3-I to LC3-II in SC79-treated cells that expressed low levels of MGST1. (d) ATG16L1 expression and the conversion of LC3-I to LC3-II in MK2206-treated cells that expressed high levels of MGST1. n = 3 in each group. *p < .05, compared with control group; #p < .05, compared with RNAi-MGST1 or MGST1 group.
MGST1-mediated ferroptosis by regulating the Akt/GSK-3β pathway
To analyze the effect of Akt/GSK-3β signal activation on MGST1-mediated ferroptosis, SGC7901 cells that expressed high or low levels of MGST1 were treated with MK-2206 and SC79, respectively. As shown in Figure 6(a) to (e), the effects of MGST1 overexpression on cell viability and ferroptosis events were antagonized by MK-2206. Similarly, the knockout of MGST1 induced a reduction in cell viability and GSH levels along with an increase in MDA, iron, and ROS levels; these effects were reversed by SC79 treatment (Figure 6(f) to (j)). MGST1 inhibited ferroptosis by inducing the Akt/GSK-3β pathway. (a) Cell viability, (b) MDA, (c) iron, (d) GSH, (e) ROS levels in MK2206-treated SGC7901 cells overexpressing MGST1. (f) Cell viability, (g) MDA, (h) iron, (i) GSH, and (j) ROS levels in SC79-treated SGC7901 cells expressing low levels of MGST1. n = 3 in each group. *p < .05, compared with control group; #p < .05, compared with RNAi-MGST1 or MGST1 group.
Discussion
Ferroptosis is a newly verified form of modulated cell death that is characterized by lipid peroxidation. 5 Further exploration of the function of ferroptosis in the progression of GC has provided novel opportunities for diagnosis and treatment. 35 Zhang et al. recently reported that cancer-associated fibroblasts (CAFs) repress ferroptosis and induce acquired chemo-resistance in GC. 36 In another study, Lee et al. demonstrated that the polyunsaturated fatty acid biosynthesis pathway potentially represents a marker for predicting the efficacy of ferroptosis-mediated cancer therapy in GC. 37 Consistent with previous observations, we found that inducers of ferroptosis could inhibit the proliferation of SGC7901 cells by promoting ferroptosis (Figure 1).
In the present study, we first identified MGST1 as a new modulator of ferroptosis in gastric carcinoma. MGST1 is expressed ubiquitously in human tissues; abnormal expression and genetic mutation of MGST1 are known to be closely associated with human disorders, such as pseudoexfoliation syndrome, macular degeneration and the initiation of cancer. 38 In our current research, we found that the expression levels of MGST1 protein and mRNA were significantly reduced in sorafenib-, erastin- and RSL3-induced ferroptosis. Moreover, all three ferroptosis inducers caused a drastic decrease in MGST1 protein; however, mRNA levels did not show a similar level of decline. The reason is that ferroptosis inducers inhibited MGST1 at transcript level, but not protein level; and MGST1 may be degraded by autophagy activation. 34 It probably indicates ferroptosis inducers affect the stability of MGST1 protein with a significant effect. The upregulation of MGST1 resulted in resistance to ferroptosis while its depletion contributed to classical ferroptotic events. Notably, the overexpression of MGST1 appeared to repress the activation of autophagy by inhibiting the expression of ATG16L1; this may represent the potential mechanism responsible for MGST1-triggered ferroptosis resistance. As expected, high expression levels of ATG16L1 partially reversed the inhibitory effect of MGST1 overexpression on ferroptosis. It is well known that the activity of the conserved ATG12-ATG5-ATG16L1 complex is crucial for the conversion of LC3-II and the formation of autophagosomes. 34 Our results suggested that the overexpression of MGST1 significantly inhibited the formation of the ATG12-ATG5-ATG16L1 protein complex while RSL3 treatment antagonized this effect in SGC7901 cells (Figure 2). In addition, the high expression of MGST1 markedly reduced the conversion of LC3-I to LC3-II (Figure 3). In contrast, RSL3 treatment significantly induced LC3-II conversion in SGC7901 cells. Therefore, we showed that the repression of ferroptosis by MGST1 overexpression was related to the inactivation of autophagy. Although more experiments are needed to investigate the specific regulatory mechanism responsible for the role of MGST1 in ferroptosis in SGC7901 cells, our results highlighted a novel function of MGST1 in autophagy in addition to the modulation of inflammation, proliferation, and apoptosis.
Increasing evidence highlights the importance of autophagy as a novel mechanism of ferroptosis and also provides new insights regarding regulated cell death.39,40 Mechanistically, selective autophagy (namely ferritinophagy) could lead to ferroptosis by regulating the degradation of ferritin. For example, dihydroartemisinin represses growth and stimulates ferroptosis in leukemia cells via the autophagy-dependent degradation of ferritin. 41 In addition, some researchers recently reported that the activation of ferritinophagy is required for Akt signaling to modulate ferroptosis in GC cells. 42 Based on previous observations, the present findings revealed that MGST1 protected against ferroptosis by modulating the autophagy signaling pathway in SGC7901 cells. Moreover, activation of the Akt/GSK-3β pathway is involved in regulating autophagy. 43 Akt signaling has also been also found to modulate the expression of autophagy flux proteins, such as Beclin1, Atg5, Atg12, Atg16L1, Atg7 and Atg3. 44
Emerging evidence suggests that the Akt/GSK-3β pathway is widely activated in many malignant tumors. 20 In addition, studies have reported that the Akt/GSK-3β axis plays an important role in inducing cell growth and repressing apoptosis in GC cells.20,45 MGST1 has also been shown to modulate the activity of Akt, the fundamental regulator of the Akt/GSK-3β pathway. 20 Thus, we hypothesized that MGST1 may promote autophagy-dependent ferroptosis resistance in GC cells by activating the Akt/GSK-3β pathway. Consistent with previous results, we showed that the overexpression of MGST1 induced the inactivation of autophagy by activating the Akt/GSK-3β signal pathway (Figures 5 and 6), whereas its depletion had the opposite effects. LC3-II is the downstream effector of the Akt/GSK-3β signaling pathway and promotes autophagy to regulate cancer cell survival and tumor occurrence. 46 In this study, we also found that MGST1 inhibited ATG16L1 expression and the conversion of LC3-I to LC3-II by activating Akt/GSK-3β axis.
Conclusion
Collectively, our findings indicated that the regulatory effect of MGST1 on ATG16L1 expression serves as an important event that represses the activation of autophagy, restricts autophagic ferritin degradation, and eventually leads to resistance to ferroptosis in GC cells. The overexpression of MGST1 induces cell proliferation by activating the Akt/GSK-3β signal pathway.
Supplemental Material
Supplemental Material - Microsomal glutathione S-transferase 1 targets the autophagy signaling pathway to suppress ferroptosis in gastric carcinoma cells
Supplemental Material for Microsomal glutathione S-transferase 1 targets the autophagy signaling pathway to suppress ferroptosis in gastric carcinoma cells by Z Peng and N Peng in Human & Experimental Toxicology
Footnotes
Acknowledgements
The authors thank the Hubei Key Laboratory of Kidney Disease Pathogenesis and Intervention.
Author contributions
ZTP and NP conceived and designed the research. NP conducted the experiments. ZTP analyzed and interpreted the data. NP drafted the manuscript. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.