
Abstract
Arsenic trioxide (ATO) has been found to be effective in acute promyelocytic leukemia. However, ATO-induced severe cardiotoxicity limits its clinical application. To date, the mechanisms of ATO-induced cardiotoxicity remain unclear. It is hypothesized that ferroptosis may trigger ATO-induced cardiotoxicity; however, this has not yet been investigated. To clarify this hypothesis, rat cardiomyocyte H9c2 cells were treated with ATO with or without ferrostain-1 (Fer-1). The results indicated that ATO exposure induced H9c2 cell death and apoptosis, and the ferroptosis inhibitor Fer-1, administered for 24 h before ATO exposure, suppressed ATO-induced cell death, and apoptosis, as determined by Annexin V-APC/7-AAD apoptosis assay. Furthermore, Fer-1 displayed a cardioprotective effect through inhibiting the ATO-induced production of intracellular reactive oxygen species, improving the ATO-induced loss of the mitochondrial membrane potential, alleviating hyperactive endoplasmic reticulum stress, and alleviating the ATO-induced impairment in autophagy in H9c2 cells. Overall, the cardioprotective effect of Fer-1 against ATO-induced cell injury implies that ATO may trigger ferroptosis to induce cardiotoxicity. These findings lay the foundation for exploring the potential value of ferroptosis inhibitors against ATO-induced cardiotoxicity in the future.
Introduction
Arsenic trioxide (ATO), a traditional Chinese medicine, has been found to be effective in acute promyelocytic leukemia.1-3 However, ATO-induced cardiotoxicity, including QT prolongation, torsades de pointes, and sudden cardiac death, limits its clinical application. 4 The mechanism of ATO-induced cardiotoxicity mainly involves accumulation of reactive oxygen species (ROS) and intracellular calcium overload.5,6 Increasing evidence indicates that ATO is able to trigger cardiomyocytes primarily by apoptosis, autophagy, and necrosis.7,8 However, the detailed mechanisms underlying the cardiotoxicity of ATO remain unclear. Further, although several studies have demonstrated that the endogenous ingredient metallothionein 9 and natural compounds such as resveratrol, 10 salvianolic acid A, 11 ascorbic acid-tocopherol combination, 12 and eugenol 13 have protective effects against ATO-induced cardiotoxicity, currently, there is no effective clinical treatment for ATO-induced cardiotoxicity.
Preventing cardiac cell death is recommended as an effective cardioprotective strategy. 14 It has long been accepted that caspase-dependent apoptosis is the major type of regulated cell death in cardiomyocytes. 7 Nonetheless, autophagy and necrosis have also been implicated in cardiac injury,7,8 in which ROS are an important interaction point; thus, ROS are expected to be a target to prevent ATO-induced cardiotoxicity. 15 Ferroptosis, characterized morphologically by the disappearance of mitochondria crista and rupture of mitochondria membranes, is a type of iron and ROS-dependent cell death. 16 Intriguingly, ATO was reported to induce excessive ROS generation and lipid peroxidation, leading to mitochondrial dysfunction. 11 Ferroptosis has also been newly characterized as a form of arsenite-induced neuronal cell death. 17 Hence, ATO may trigger cardiomyocyte cell death by ferroptosis. However, whether ATO induces ferroptosis in cardiomyocytes remains unknown. Therefore, in this study, rat cardiomyocytes (H9c2 cells) were treated with the ferroptosis inhibitor ferrostain-1 (Fer-1) to study the effect of Fer-1 against ATO-induced cardiotoxicity.
Methods
Cell culture
H9c2 cells were acquired from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS; Gibco, USA) at 37°C, in 5% CO2. ATO and Fer-1 were purchased from Harbin YI-DA Pharmaceutical Ltd. (Harbin, China) and Selleck (Shanghai, China), respectively.
Cell viability assay
After 24 h of treatment with different doses of ATO, 10 μL of CCK-8 reagent (Applygen, Beijing, China) was added to 100 μL DMEM to obtain a working solution. After washing the cells with the original medium, the working solution was added, and the cells were incubated for 2 h at 37°C. Then, the absorbance was measured at 450 nm by a Thermo Scientific Multiskan GO microplate reader.
7-AAD (7-amino-actinomycin D) is a nucleic acid dye that cannot pass through the normal plasma membrane. However, the permeability of the plasma membrane to 7-AAD gradually increases with cell apoptosis and cell death, and 7-AAD combined with the controlled degradation of DNA can emit bright red fluorescence at a suitable wavelength. Thus, cells with a red fluorescence are considered dead cells. Here, H9c2 cells were pretreated with 32 μM Fer-1 for 24 h, and then 15 μM of ATO was added and cell treatment continued for another 24 h. After the drug treatments, the cells were washed with phosphate buffered saline (PBS), and 500 μL of binding buffer with 5 μL 7-AAD dye solution were added. Images were taken with a fluorescence microscope (ZEISS Axio Observer D1, Germany) in a blinded manner.
Annexin V-APC/7-AAD apoptosis assay
An Annexin V-APC/7-AAD apoptosis kit (KeyGEN BioTECH, China) for flow cytometry was used to detect the early apoptosis and necrosis of cells. Cells were pretreated with 32 μM Fer-1 for 24 h, and then 15 μM of ATO was added and the cells were treated for another 24 h. Next, the cells were harvested, washed twice with cold PBS, and incubated with 5 μL of Annexin V-APC and 5 μL of 7-AAD in 500 μL binding buffer for 10 min in the dark at room temperature. A flow cytometer (BD Biosciences, USA) was used to detect the cellular fluorescence.
Oxidative stress detection
ROS generation was detected by DCFH-DA staining (Beyotime Biotechnology, China). After washing with PBS, the cells were incubated with DCFH-DA (10 μmol/L) at 37°C in the dark for 30 min. Fluorescent images were captured with a fluorescence microscope (ZEISS Axio Observer D1, Germany) in a blinded manner. To further assess the oxidative stress level, the content of malondialdehyde (MDA) was measured by a commercially available kit (Beyotime Biotechnology, China).
Detection of mitochondrial membrane potential by JC-1
Mitochondrial transmembrane potential changes were detected by JC-1 (KeyGEN BioTECH, China), a lipophilic cation that can selectively enter mitochondria. H9c2 cells were incubated with JC-1 at 37°C in the dark for 20 min. Then, the loading solution was replaced with fresh medium and the fluorescence signal was captured by fluorescence microscopy. JC-1 as a monomer in the cytoplasm emitting green fluorescence can be rapidly absorbed into the mitochondria as a polymer emitting red fluorescence, depending on the polarity of the membrane potential. Consequently, a decrease in the red/green fluorescence intensity ratio suggests mitochondrial depolarization.
Transmission electron microscopy
Mitochondria changes caused by ATO and Fer-1 were observed by transmission electron microscopy (TEM). Fixative for TEM (Servicebio, China) was used to fix the cells at room temperature for 5 min. Then, the cells were gently scraped in one direction using a cell scraper and sucked into a 1.5 ml centrifuge tube. Next, 300 g of cells were centrifuged for about 2°min. New fixative for TEM was added to suspend the cell cluster and the cells were further fixed at room temperature in the dark for 30 min. The samples were stored at 4°C and transported to Wuhan Servicebio Technology Co., Ltd for TEM.
Quantitative real-time polymerase chain reaction
Total RNA was extracted from the H9c2 cells using Trizol reagent (Invitrogen, USA). The target genes were quantified by Power SYBR™ Green PCR (Thermo Fisher Scientific, USA). The primer sequences of glutathione peroxidase 4 (GPX-4) were: F:5′-GCCGAGTGTGGTTTACGAAT-3′, R: 5′- GGCTGGACTTTCATCCATTT-3; the primer of GAPDH was purchased from Sangon Biotech. The results were analyzed using the 2−ΔΔCt method.
Western blot analysis
Total proteins were loaded by 10% SDS-PAGE, transferred to polyvinylidene difluoride membranes, and blocked with 5% skim milk. Membranes were incubated with the following primary antibodies at 4°C overnight: GPX-4 (#bs-3884R; Bioss, China), B-cell lymphoma 2 (Bcl-2; # T40056; Abmart, China), Bcl-2-associated X protein (Bax; #T40051; Abmart, China), cleaved caspase-3 (#9644T; Cell signaling technology, USA), DNA damage-inducible transcript 3 protein/enhancer-binding protein homologous protein (DDIT3/CHOP; #AF6684; Beyotime Biotechnology, China), Beclin 1 (#AF5123; Beyotime Biotechnology, China), SQSTM1/p62 (#AF5312; Beyotime Biotechnology, China), ATG5 (#T55766; Abmart, China), and LC3B (#T55992; Abmart, China). A digital gel image analysis system Amersham Imager 600 (GE Healthcare Bio-Sciences, USA) was used to measure and analyze the density of the bands.
Statistical analysis
Each experiment was independently repeated at least three times. The data are expressed as the mean ±standard deviation (SD). One-way ANOVA was used to compare multiple data and t-tests were used to compare data between two groups. Analyses were performed using GraphPad Prism 8.0 (SPAA, Inc., Chicago, USA). Statistical significance was defined as a p value < 0.05.
Results
Fer-1 protects against ATO-induced cell death
The toxicity of ATO in H9c2 cells was first examined. Cell viability was significantly decreased at an ATO concentration of 5 μM, and cell viability was further decreased with increasing ATO concentration (IC50=20.7 μM) (Figure 1 (a) and (b)). H9c2 cells were in a poor condition at an ATO concentration of 20 μM (IC50) and were difficult to rescue with Fer-1. Further, the cells exhibited significant changes in morphology and cell number counts at an ATO concentration of 15 μM; the cell viability at this concentration was (72±5) % (Figure 1 (c)). Thus, 15 μM ATO was used in further experiments. 7-AAD-positive cells were considered dead cells. The results indicated that there were more 7-AAD-positive cells in the ATO group while the Fer-1 treatment significantly decreased ATO-induced cell death (Figure 2 (a) and (b)). ATO-induced H9c2 cell death. (a) ATO was administered for 24 h. (b) The IC50 curve of Figure 1a. (c) The morphology and count changes of H9c2 cells when treated with ATO (10 μM, 15 μM). ATO, arsenic trioxide. *, p < 0.05 vs control group; **, p < 0.001 vs control group. Fer-1 protects against ATO-induced cell death. (a, b) H9c2 cells were pretreated with Fer-1 (32 μM) for 24 h and then exposed to ATO (15 μM) for another 24 h. 7-AAD staining showed more 7-AAD positive cells in the ATO group while Fer-1 treatment reduced ATO-induced cell death. Fer-1, ferrostain-1; ATO, arsenic trioxide. *, p < 0.05 vs control group; #, p < 0.05 vs ATO group. Scale bar is 100 μM.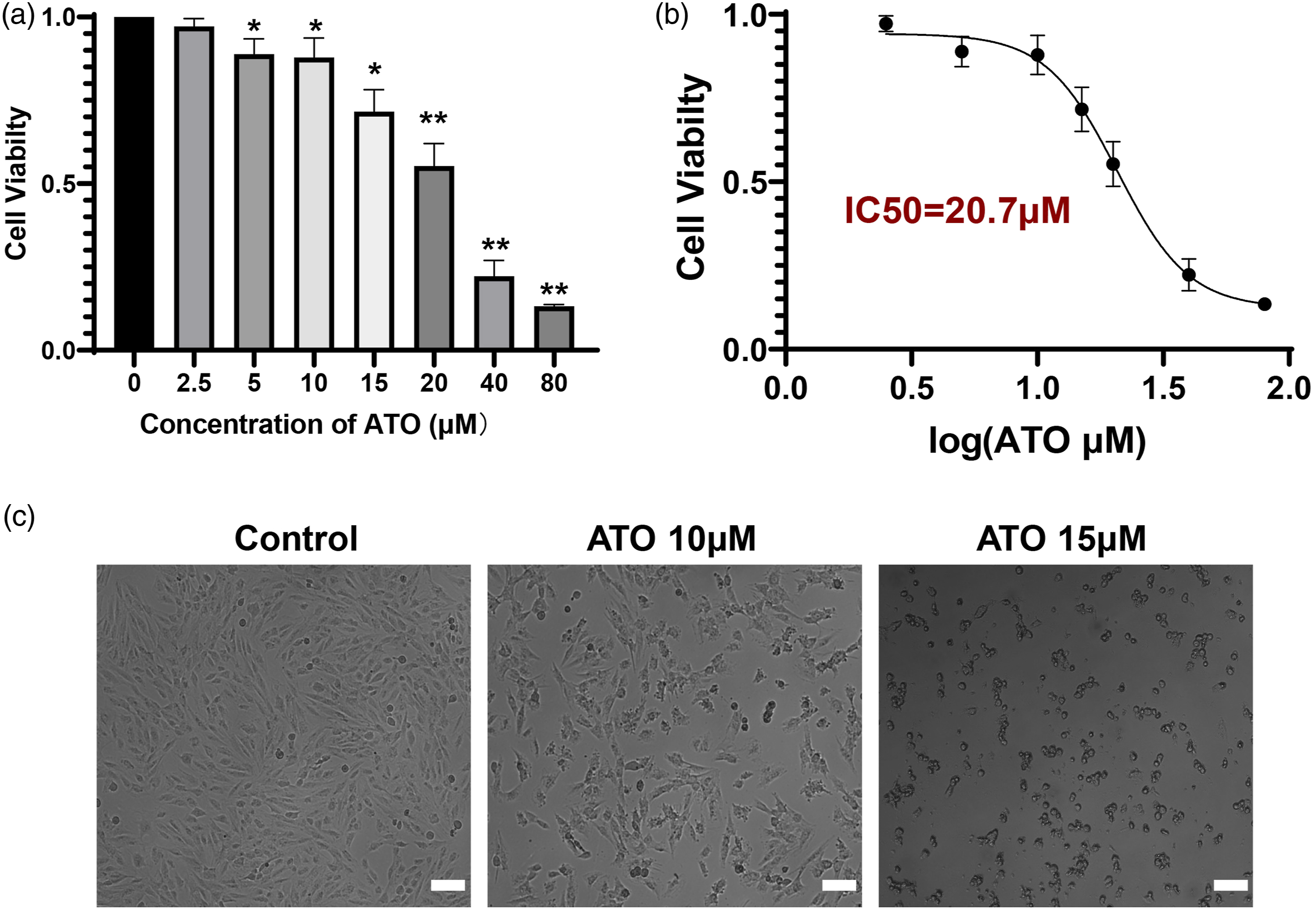

Fer-1 attenuates ATO-induced cell apoptosis
Compared with the obvious H9c2 cell apoptosis in the ATO group, a significant reduction in apoptosis was observed in the ATO+Fer-1 group (Figure 3 (a) and (b)). Further, Fer-1 treatment significantly reduced the expression of the proapoptotic Bax and cleaved caspase-3 activity but increased the expression of the anti-apoptotic Bcl-2, as compared to the ATO group (Figure 3 (c–f)). Therefore, these results suggest that Fer-1 inhibits ATO-induced early-stage apoptosis. Fer-1 attenuates ATO-induced cell apoptosis. (a, b) H9c2 cells were pretreated with Fer-1 (32 μM) for 24 h and then exposed to ATO (15 μM) for another 24 h. Cell apoptosis was detected by Annexin V-APC/7-AAD flow cytometry. (c, d, e, f) Bax (Bcl-2 associated X), Bcl-2 (B-cell lymphoma 2), and cleaved caspase-3 were detected by Western blot. Fer-1, ferrostain-1; ATO, arsenic trioxide. *, p < 0.05 vs control group; #, p < 0.05 vs ATO group.
Fer-1 suppresses ATO-induced cell oxidative stress
Compared with the control group, the levels of ROS were greater in the ATO group, and Fer-1 suppressed ATO-induced ROS production (Figure 4 (a) and (b)). ATO-stimulated cells exhibited increased MDA production (a membrane lipid peroxidation biomarker), and this was essentially relieved by Fer-1 (Figure 4 (c)). JC-1 was applied to verify whether Fer-1 protected the function of mitochondria from ATO-induced cell oxidative stress in H9c2 cells. The mitochondria of H9c2 cells in the control group were brightly stained with red fluorescence, while H9c2 cells treated with ATO exhibited fewer and less-stained mitochondria. Fer-1 pre-treatment prevented ATO-induced loss of mitochondrial membranes (Figure 4 (d) and (e)). Taken together, these findings indicate that pre-treatment with Fer-1 suppresses ATO-induced oxidative stress in H9c2 cells. Fer-1 suppresses ATO-induced cell oxidative stress. (a, b) DCFH-DA was used to detect the level of reactive oxygen species. (c) The oxidative stress biomarker MDA was measured by a commercially available kit. (d, e) JC-1 was used to measure the mitochondrial membrane potential. H9c2 cells were pretreated with Fer-1 (32 μM) for 24 h and then exposed to ATO (15 μM) for another 24 h. Fer-1, ferrostain-1; ATO, arsenic trioxide; MDA, malondialdehyde. *, p < 0.05 vs control group; #, p < 0.05 vs ATO group; orange scale bar is 200 μM; white scale bar is 100 μM.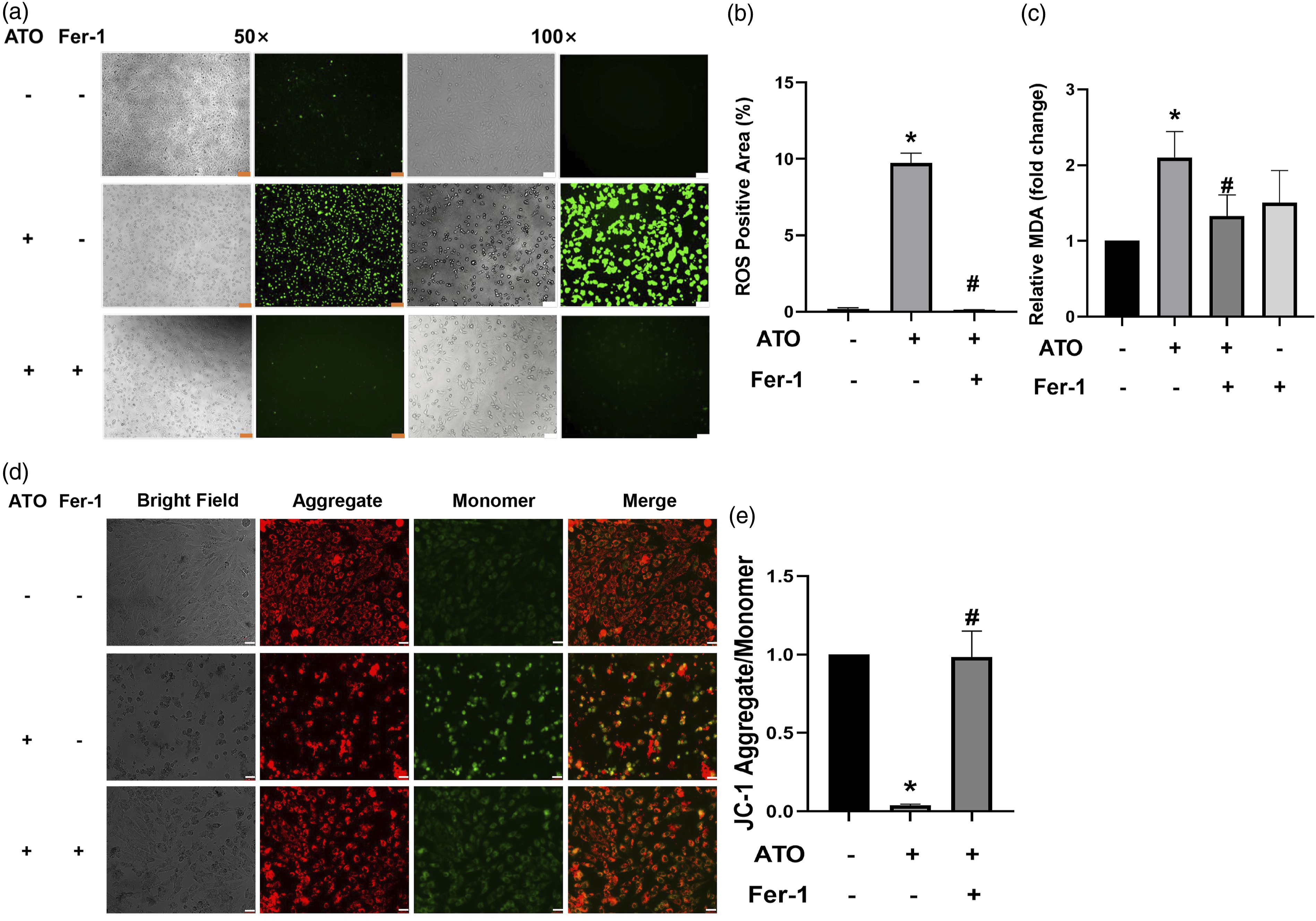
Fer-1 decreases ATO-induced of endoplasmic reticulum stress
Compared with the control group, a significant increase in the protein expression level of DDIT3/CHOP was found in the ATO group, whereas Fer-1 treatment markedly decreased the level of this protein (Figure 5 (a) and (b)). This result indicates that Fer-1 alleviates ATO-induced endoplasmic reticulum stress. Fer-1 alleviates ATO-induced endoplasmic reticulum stress and ferroptosis. H9c2 cells were pretreated with Fer-1 (32 μM) for 24 h and then exposed to ATO (15 μM) for another 24 h. (a, b) The level of CHOP was measured by Western blot. (c) Mitochondria changes caused by ATO and Fer-1 were observed through transmission electron microscopy. (d, e) The protein level of GPX-4 was detected by Western blot. (f, g) The mRNA level of GPX-4 was detected by quantitative real-time polymerase chain reaction. *, p < 0.05 vs control group; #, p < 0.05 vs ATO group. Fer-1, ferrostain-1; ATO, arsenic trioxide; CHOP, CCAAT/enhancer-binding protein; GPX-4, glutathione peroxidase 4.
Fer-1 inhibits ATO-induced ferroptosis
Mitochondria are the powerhouse of cardiomyocytes. Myocardial mitochondria in ATO-treated H9c2 cells were terribly distorted and enlarged; however, this was rescued by Fer-1 treatment (Figure 5 (c)). GPX-4 is an important protein in ferroptosis; it is the only enzyme able to directly decrease complex phospholipid hydroperoxides in the cell membrane and lipoproteins generated from free radical oxidation under oxidative stress. 18 In this study, ATO-induced a remarkable reduction in the mRNA and protein expression levels of GPX-4, whereas Fer-1 treatment markedly increased the levels of this protein (Figure 5 (d–g)). These results indicate that Fer-1 inhibits ATO-induced ferroptosis via enhancement of GPX-4.
Fer-1 alleviates the ATO-induced impairment in autophagy
To verify whether Fer-1 influences ATO-induced H9c2 cell autophagy, autophagy-related proteins were detected (Figure 6). The protein expression level of p62 was distinctly up-regulated, while Beclin 1 and ATG5 were down-regulated with ATO treatment. Fer-1 treatment effectively corrected these abnormalities (Figure 6). However, LC3B-II was only up-regulated in the Fer-1+ATO treatment but not in the ATO treatment. These results demonstrate that Fer-1 alleviates the ATO-induced impairment in autophagy. Fer-1 alleviates the ATO-induced impairment in autophagy. H9c2 cells were preincubated with Fer-1 (32 μM) for 24 h and then exposed to ATO (15 μM) for another 24 h. (a-e) The proteins of SQSTM1/p62, Beclin 1, ATG5 and LC3B were detected by Western blot. Fer-1, ferrostain-1; ATO, arsenic trioxide. *, p < 0.05 vs control group; #, p < 0.05 vs ATO group.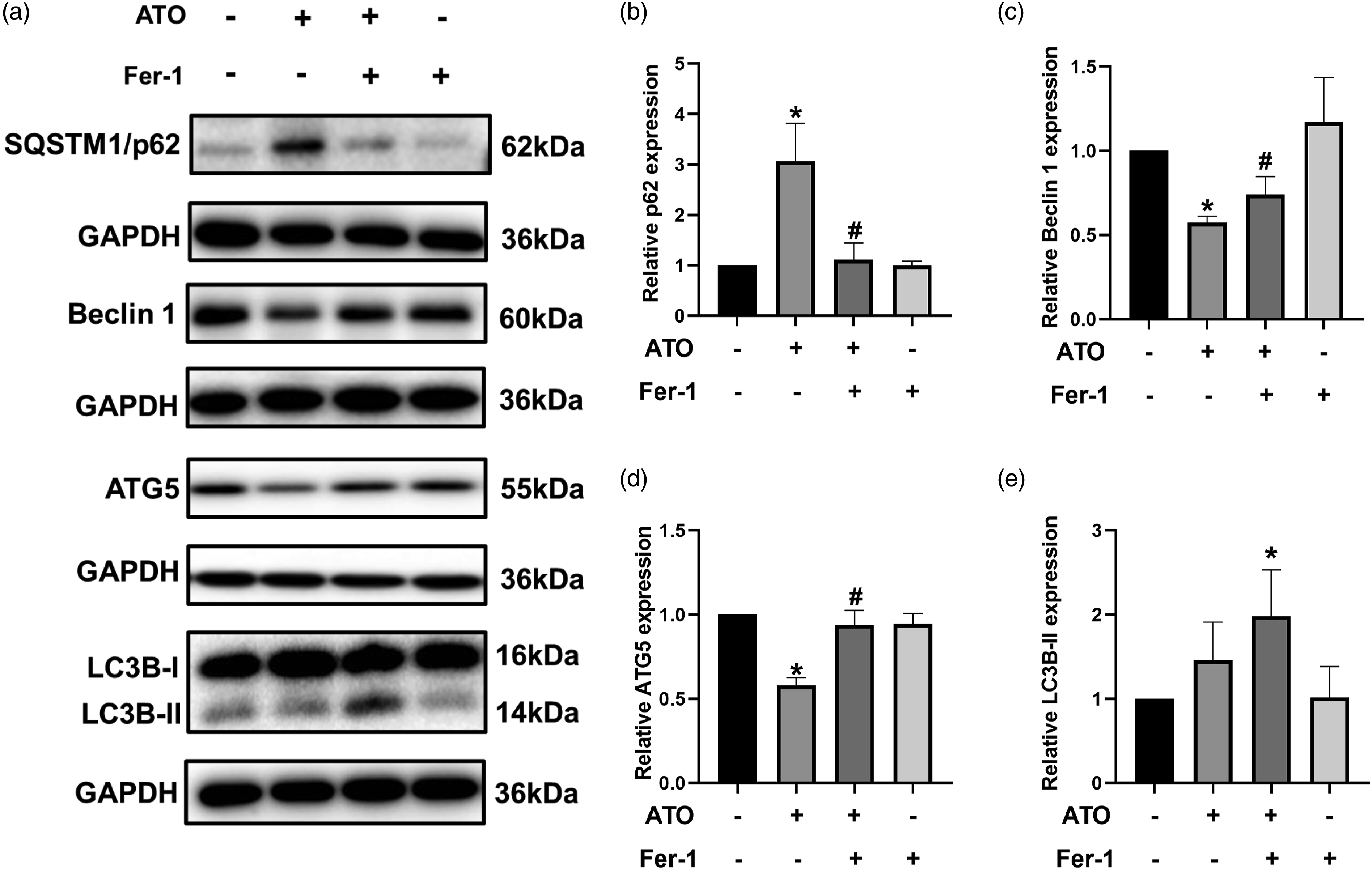
Discussion
ATO, a traditional Chinese medicine, has been found to be effective in acute promyelocytic leukemia.1-3 However, ATO-induced cardiotoxicity, including QT prolongation, torsades de pointes, and sudden cardiac death, limits its clinical application. 4 Cardiomyocyte cell death is the main mechanism of ATO-induced cardiotoxicity, 14 but the detailed mechanisms underlying cardiomyocyte cell death induced by ATO remain unclear. In this study, the results demonstrated that ATO caused H9c2 cell death by producing a marked loss of viability and inducing cell apoptosis. The ferroptosis inhibitor Fer-1 protected ATO-induced cardiomyocytes from cell death and apoptosis, which means that ferroptosis is a form of H9c2 cell death in response to ATO exposure.
Enhanced oxidative stress is the key mechanism of ATO-induced cardiotoxicity. 19 Excessive levels of ROS lead to oxidative stress, resulting in different types of cell death, including autophagy, apoptosis, and necrosis.20,21 Notably, GPX-4, an important marker of ferroptosis, 22 is the only enzyme able to directly decrease complex phospholipid hydroperoxides in the cell membrane and lipoproteins generated from free radical oxidation under oxidative stress by converting lipid hydroperoxides into non-toxic lipid alcohols. 18 In this study, it was found that ATO triggers ferroptosis in cardiomyocytes. Specifically, this mechanism involved the distortion and enlargement of myocardial mitochondria, the generation of ROS, the accumulation of MDA, and the depletion of GPX-4. Fer-1 mitigated ROS and MDA production, and also improved the synthesis of GPX-4, which was suppressed by ATO. Moreover, previous studies have demonstrated that endoplasmic reticulum stress is involved in ATO-induced heart injury,5,23 and the current results indicated that ATO also harms endoplasmic reticulum function as confirmed by the upregulation of CHOP. Fer-1 treatment may have a protective effect against ATO-induced endoplasmic reticulum stress.
Autophagy is regarded as the ultimate consequence of metal-induced oxidative stress, 24 playing an important role in homeostasis and the function of the heart. 25 Current evidence indicates that ferroptosis is also a type of autophagy-dependent cell death. 26 However, a previous study only found a slight change in autophagy in ATO-induced H9c2 cell injury; this may be because this study only used CytoID green fluorescent probes to detect the level of autophagy. 27 In the current study, the ferroptosis inhibitor Fer-1 was found to protect against ATO-induced H9c2 cell death via the autophagy pathway. Autophagy-related proteins such as SQSTM1/p62, Beclin 1, ATG5, and LC3B were detected in the current study. Interestingly, in contrast to other studies of autophagy,28,29 ATG5 and Beclin1 were down-regulated while p62 was up-regulated in the ATO group, and these changes were alleviated by Fer-1. However, LC3B-II was only significantly up-regulated in the Fer-1+ATO group, which means that ATO may induce impairment in autophagy by inducing the accumulation of oxidative stress while Fer-1 may activate autophagy or alleviate the impairment in response to oxidative stress in order to protect the cells from apoptosis. 30
A limitation of this study is that only the H9c2 cell line was used and there were no animal experiments performed. In the future, we intend to confirm the mechanism of Fer-1 protection against ATO-induced cardiomyocyte injury in vivo.
In summary, the ferroptosis inhibitor Fer-1 protects H9c2 cells from ATO-induced cell death, apoptosis, endoplasmic reticulum stress, and autophagy. This study suggests that the inhibition of ferroptosis in cardiomyocytes may be a new potential way to treat ATO-induced cardiotoxicity. These findings lay the foundation for exploring the potential value of Fer-1 against ATO-induced cardiotoxicity in the future.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC) (No. 82070154), National Science and Technology Key Projects (No. 2017ZX09304029004), and Children’s Medicine Research Project of Beijing Children’s Hospital, Capital Medical University (No. YZZD202006).