
Abstract
Acrylamide (ACR) is a water-soluble chemical applied in industrial and laboratory processes. The neurotoxicity induced by acrylamide involves both peripheral and central nervous system. Hence, there is a growing urgency to investigate the mechanisms of acrylamide-induced neurotoxicity and search novel therapeutic target for the nerve repair. The effects of ACR on the proliferation, reactive oxygen species (ROS) and iron production of dorsal root ganglia (DRG) neurons and Schwann cells were determined. 5-Ethynyl-2′-deoxyuridine (EDU) staining and transwell assay were applied to detect the proliferation and migration capacity of DRG cells. Ferrostatin-1 (Fer-1) was used to suppress ferroptosis induced by ACR. RT-PCR analysis was performed to examine the expression of neurotrophic factors including brain-derived neurotrophic factor (BDNF), nerve growth factor (NGF), vascular endothelial growth factor (VEGF) and glial cell line-derived neurotrophic factor (GDNF). Moreover, Iron, ROS, malondialdehyde (MDA) and glutathione (GSH) contents were measured to reveal the regulation of ferroptosis in ACR-related nerve injury. ACR inhibited the proliferation and migration of DRG neurons and the supplementation of Fer-1 reversed the effects induced by ACR. Besides, the treatment of Fer-1 effectively increased the expression of NGF, BDNF, VEGF and GDNF. Furthermore, ACR increased the iron level, MDA and ROS contents while inhibited the level of GSH. It was unveiled that ACR attenuated the proliferation, migration and neuron repair of DRG neurons through regulating ferroptosis. The modulation of ferroptosis might be a promising therapeutic strategy and provide references for future treatment of acrylamide-induced nerve damage.
Introduction
Acrylamide (ACR) is a water-soluble monomer used in various chemical and industrial yields such as the production of cosmetics, plastics and toiletries, among others.1,2 Besides, acrylamide is also generated during the food processing at high temperatures. 3 The Scientific Committee on Toxicity Ecotoxicity and Environment declared the neurotoxicity, genotoxicity and reproductive toxicity of acrylamide in humans and animals in 2001. 4 Subchronic exposure to acrylamide causes sensory, motor and autonomic dysfunctions such as ataxia, gait abnormalities, skeletal muscle weakness and numbness of hands and feet.5,6 Given the exposure to acrylamide is inevitable, it is crucial to search new treatment strategies for the repair of acrylamide-induced nerve injury and functional reconstruction.
Ferroptosis is a novel type of regulated cell death induced by the iron accumulation and lipid peroxidation. 7 The disruption of lipid peroxide reduction systems causes the accumulation of reactive oxygen species (ROS), which could be induced by destroying the role of the system xCT (SLC7A11) or glutathione peroxidase 4 (GPX4).8,9Recent findings have unveiled that ferroptosis involved that various pathophysiological processed and disorders including cancers,10,11 neurodegenerative disorders 12 and cardiovascular diseases. 13 The expression of ACSL4 up-regulated in early brain injury (EBI) and brain damage after subarachnoid hemorrhage (SAH). 14 ACSL4 induced ferroptosis and promoted brain damage through mediating lipid metabolism. Menon et al. identified that up-regulation of heme oxygenase 1 (HMOX1) triggered ferroptosis in Sickle cell disease, which resulted in cardiomyopathy. 15 Furthermore, the application of ferroptosis inhibitors inhibited cardiomyopathy while ferroptosis inducers accelerated cardiac injury. However, whether ferroptosis is involved in the ACR-induced neural damage remains largely unclear.
In the present study, it was firstly determined that the ferroptosis induced by ACR mainly occurred in DRG neurons rather than Schwann cells. ACR inhibited the cell proliferation and migration ability and increased the iron, MDA and ROS concentration. The protective role of Fer-1 on ACR-prompted neuropathy by alleviation of ferroptosis and enhancement of neurotrophic factors. Hence, Inhibition of Ferroptosis might be a novel potential treatment strategy for ACR-induced neurotoxicity.
Materials and methods
Cell culture
The DRG neurons were isolated and purified from L1-L6 DRGs of 3-weeks-old male Sprague-Dawley rats (Cavens Lab Animal Co. Changzhou, China) and cultured as previously reported.16,17 The DRG neurons were cultured in neurobasal medium (Gibco, USA) consisting of 10 μM Ara-C (Sigma, China) and 2% B27 supplement (Gibco, USA) at 37°C in the incubator with 5% CO2. The Schwann cells were prepared from L1-L6 DRGs of 3-weeks-old male Sprague-Dawley rats (Cavens Lab Animal Co. Changzhou, China) as described previously.18,19 The purified DRG neurons and Schwann cells were identified with immunofluorescence staining using GAP-43 (Affinity, China) and S-100β antibody (Affinity, China), respectively. Acrylamide (ACR, 1mmlol/L) was used to induce neurotoxicity and nerve damage to DRG neurons and Schwann cells as previously described. 20
Cell proliferation assay
Cell proliferation was evaluated using CCK-8 (CCK-8, Beyotime, China) and EDU (RiboBio, China) incorporation assays. Briefly, 2x104 cells of DRG neurons or Schwann cells were seeded into 96-well plates in triplicate after ACR treatment. After incubation at 37°C for 2 h, CCK-8 solution (10 μL) was added to each well and the absorbance of 450 nm was verified with a microplate Reader (Thermo Fisher Scientific, USA). For the EDU assay, 50 mM EDU solution was added to the culture medium and incubated for another 2 h. Then the cells were fixed with 4% paraformaldehyde and stained with Apollo dye solution for proliferating cells. Besides, nucleic acids were stained with DAPI. The proliferation rate was the ratio of the EDU-positive cells to the number of DAPI-stained cells.
Transwell assay
The cell migration assay was carried out using a transwell chamber of 8 μm pore filters (Corning, USA). Firstly, DRG neurons (at a density of 1 × 105) were collected and incubated in the upper chamber with 100 μL serum-free medium. 600 μL medium supplemented with 10% FBS was added to the lower chamber. After culture for another 24 h, Cotton swab was used to remove the non-metastatic DRG neurons. And the migrated cells on the surfaces of lower chambers were fixed and stained using 500 μL crystal violet (Beyotime, China) for 5 min. The stained cells were washed with fresh PBS for three times and then counted in three random fields under a microscope (Leica, Germany).
Measurement of reactive oxygen species (ROS)
ROS production of DRG neurons and Schwann cells were assessed using DCFH-DA (Beyotime, China) after exposure to ACR. Briefly. a total of 4 × 104 of cells were collected and incubated with 10 μM DCFH-DA working solution at 37°C for 30 min. The cell pellet was collected by centrifugation at 10k rpm for 5 min to remove DCFH-DA. Then the cells were resuspended in fresh PBS and the fluorescence intensity was detected at excitation and emission wavelengths of 488 nm and 525 nm using a flow cytometer (FACSCalibur, BD). The results were represented as average fluorescence intensity.
Intracellular iron assay
The relative intracellular iron levels of DRG neurons and Schwann cells were measured using the Iron assay kit (Abcam, USA) according to the manufacturer’s instructions. 1 × 106 cells after indicated treatment were collected and homogenized in iron assay buffer. Then the cells were centrifuged at 16,000 × g for 10 min and 90 μL buffer was added to 10 μL supernatant. Moreover, 5 μL iron reducer was added and followed by incubation for 25 min. Finally, the iron concentrations were detected with 100 μL iron probe under a microplate reader at a wavelength of 593 nm.
Detection of cysteine, malondialdehyde (MDA) and glutathione (GSH) levels
The concentrations of cysteine, MDA and GSH were assessed using Cysteine Colorimetric assay kit (Elabscience, China), Lipid Peroxidation MDA assay kit (Beyotime, China) and total glutathione/Oxidized glutathione assay kit (Nanjing Jiancheng Bioengineering Institute, China) according to the manufacturer’s instructions.
Realtime quantitative PCR analysis
Total RNA was isolated from DRG neurons or Schwann cells through TRIzol reagent (Invitrogen, CA, USA). cDNA was derived from 1 μg RNA using SuperScript III Reverse Transcriptase (Invitrogen, CA, USA). Quantitative qPCR was performed with a SYBR-Green Realtime PCR Master Mix (ToyoBo Life Sciences) in a CFX Real-Time System (Bio-Rad). PCR reaction conditions were 95°C for 3 min, followed by 40 cycles of amplification (95°C for 30 s, 55°C for 20 s, 72°C for 20 s). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA was used to normalize RNA levels. The quantification of the relative levels of BDNF, NGF, VEGF, GDNF, SLC7A11 and GPX4 were performed using 2−ΔΔCt method. The primers were synthesized by Sangon Biotech (Shanghai, China) and listed in Supplement Table S1.
Statistics analysis
All data were represented as mean ± standard deviation (SD) and analyzed using GraphPad prism 7.0 version (GraphPad Software, CA, USA). Difference between means was determined by one-way ANOVA and subsequent Tukey’s post-hoc test for multiple groups or 2-tailed independent Student t-test for the significance between two groups. p < 0.05 was considered as statistically significant.
Results
ACR suppressed the cell viability of DRG neurons and Schwann cells
To explore the neurotoxicity of ACR, the primary DRG neurons and Schwann cells were isolated, purified and cultured from L1-L6 Dorsal root ganglion and sciatic nerves, respectively. Primary DRG neurons were then identified through immunofluorescence staining of the specific marker GAP-43 (Figure 1(a)). Besides, the purity of Schwann cells was confirmed by immunofluorescence staining of S-100β (Figure 1(b)). Moreover, DRG neurons and Schwann cells were challenged with ACR (1mmlol/L) for 48 h to perform CCK-8 assay. As shown in Figure 1(c), ACR treatment inhibited the cell viability of both DRG neurons and Schwann cells, which confirmed the neurotoxicity induced by ACR. ACR inhibited the cell viability of DRG neurons and Schwann cells. (a) Primary DRG neurons were identified with immunofluorescence staining of GAP-43 (n = 3). Scale barr = 20 μm. (b) Immunofluorescence staining of S-100β was used to identify Schwann cells (n = 3). Scale barr = 20 μm. (c) CCK-8 assay was carried out to evaluate the cell proliferation of DRG neurons and Schwann cells after ACR treatment (1mmlol/L) for 48 h (n = 3). **p < .01 in 2-tailed t-test (c). Data are presented as mean ± SD. Abbreviations: ACR–acrylamide; DRG–dorsal root ganglia.
Ferroptosis induced by ACR mainly occurred in DRG neurons rather than Schwann cells
To investigate whether ACR caused neurotoxicity through inducing ferroptosis, the ROS and iron accumulation levels were detected using DCFH-DA probes and iron assay kit, respectively. ROS and iron accumulation are major hallmarks of ferroptosis.
21
The high ROS content induces cell death. As shown in Figures 2(a) and (b), ACR treatment promoted the ROS accumulation both in DRG neurons and Schwann cells. Furthermore, the DRG neurons after ACR treatment had higher ROS concentration than Schwann cells. Moreover, it was found that ACR triggered iron accumulation in DRG neurons, but not in Schwann cells (Figure 2(c)). Ferroptosis is a novel type of iron-dependent cell death. Therefore, it seemed DRG neurons occurred ferroptosis rather than Schwann cells after ACR treatment. SLC7A11, a subunit of xCT, and GPX4 are considered as key anti-ferroptosis proteins.
22
SLC7A11 serves as in inverse ferroptosis modulator which keeps a homeostatic redox state.
23
GPX4, a glutathione peroxidase, convert toxic hydroperoxides into lipid alcohols, thus attenuating ferroptosis.
24
As shown in Figure 2(d), the mRNA expression of GPX4 and SLC7A11 remarkably decreased in DRG neurons after ACR treatment. However, the expression of SLC7A11 was not significantly altered in ACR-treated Schwann cells. Based on the above results, it was hypothesized that ACR induced neurotoxicity through triggering ferroptosis in DRG neurons. Ferroptosis induced by ACR mainly occurred in DRG neurons rather than Schwann cells. (a, b) Flow analysis was detected the examine the ROS contents of DRG neurons and Schwann cells after ACR treatment (n = 23). (c) The relative intracellular iron levels of DRG neurons and Schwann cells were measured by the Iron assay kit (n = 3). (d) The mRNA expression of Glutathione peroxidase 4 and SLC7A11 were validated using RT-PCR analysis (n = 3). Glyceraldehyde 3-phosphate dehydrogenase mRNA was used to normalize the relative gene expression. *p < 0.05, **p < 0.01, ns = non-significant in 2-tailed t-test (b, c, d). Data are presented as mean ± SD. Abbreviations: ACR–acrylamide; DRG–dorsal root ganglia.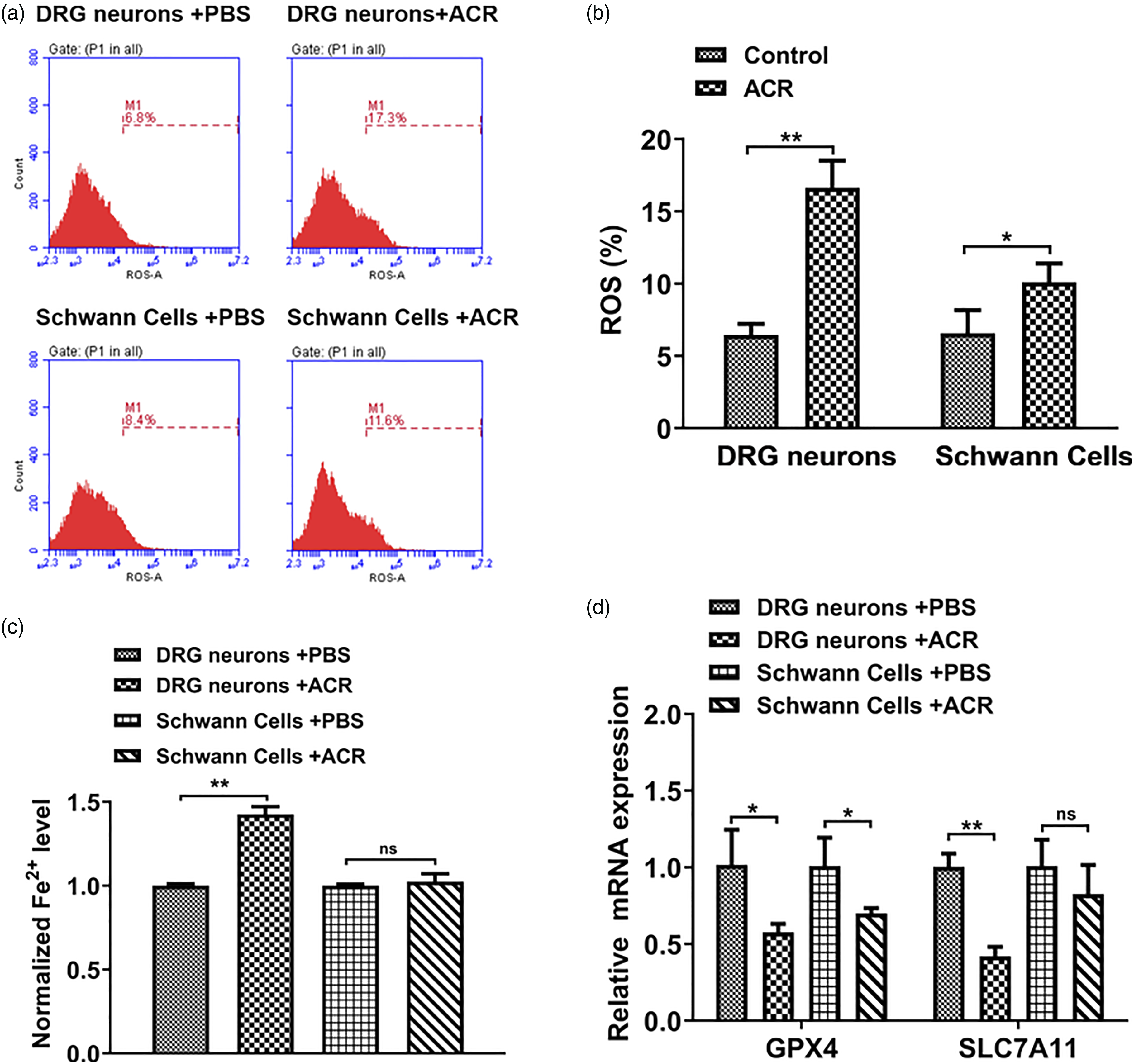
Fer-1 remarkably reversed the ACR-induced modulation of cell proliferation and migration on DRG neurons
Ferroptosis-specific inhibitor ferrostatin (Fer-1), a specific ferroptosis inhibitor, was used to suppress ferroptosis in DRG neurons. The EDU staining was performed to examine cell proliferation and the result (Figures 3(a) and (b)) revealed that Fer-1 could effectively reverse the inhibition of ACR on DRG neurons’ proliferation. Hereafter, cell migration was determined using transwell assay. As shown in Figures 3(c) and (d), ACR apparently suppressed cell migration ability of DRG neurons. However, Fer-1 treatment turned over the inhibitory role induced by ACR on DRG neurons. Fer-1 remarkably reversed the ACR-induced modulation of cell proliferation and migration on DRG neurons. (a, b) The cell proliferation of DRG neurons was quantified with EDU staining (n = 3). The EDU-labeled (red) indicated proliferation active cells and whole DRG neurons were stained with DAPI in blue. Scale barr = 20 μm. (c, d) The cell migration ability of DRG neurons was determined by Transwell assay after indicated treatment (n = 3). Scale barr = 20 μm. **p < 0.01 in Tukey’s post-hoc comparisons test (b, d). Data are presented as mean ± SD. Abbreviations: ACR–acrylamide; DRG–dorsal root ganglia; EDU–Ethynyl-2′-deoxyuridine.
Fer-1 effectively promoted neurotrophic factors expression of DRG neurons
Further investigation was carried out to determine whether Fer-1 had neurorestorative effects against ACR-induced cell death. Previous reports have identified that neurotrophic factors such as NGF, BDNF, VEGF and GDNF are involved in the repair of injured peripheral nerves.
25
The real-time PCR analysis was used to to detect the expression changes of neurotrophic factors. The results (Figures 4(a)–(d)) indicated the mRNA expression of NGF, BDNF, VEGF and GDNF obviously increased after ACR induction compared with control group. It was speculated that this phenomenon resulted from the cells attempt at self-protection against ACR-induced neurotoxic damage. Besides, Fer-1 further contributed the expressions of NGF, BDNF, VEGF and GDNF, which suggested a neuroprotective effect of Fer-1 against ACR toxicity. Fer-1 effectively promoted nerve regeneration of DRG neurons. The mRNA expression of nerve growth factor (a), Brain-derived neurotrophic factor (b), Vascular endothelial growth factor (c) and Glial cell line-derived neurotrophic factor (d) were evaluated by RT-PCR assay in DRG neurons after ACR treatment or co-treatment (ACR+Fer-1) (n = 3). Data were normalized to the reference gene glyceraldehyde 3-phosphate dehydrogenase. **p < 0.01 in Tukey’s post-hoc comparisons test (a, b, c, d). Data are presented as mean ± SD. Abbreviations: ACR–acrylamide; DRG–dorsal root ganglia.
Fer-1 inhibited ferroptosis induced by ACR in DRG neurons
Ferroptosis is characterized by the excessive accumulation of iron, the high level of intracellular ROS through the Fenton reaction and the depletion of GSH.
26
GSH protects lipid membrane through reacting with ROS to generate oxidized glutathione.
27
Besides, ROS degrades lipid and generates MDA, so the MDA level is a key intermediate product during the lipid peroxidation process.
28
Hence, it was assessed the levels of iron, ROS, GSH and MDA of DRG neurons after ACR induction or combination treatment (ACR+ Fer-1). And as shown in Figures 5(a)–(d), the iron, ROS and MDA contents were significantly elevated in the ACR-induced group, while GSH level apparently attenuated. Moreover, Fer-1 treatment effectively inhibited the ACR-induced iron, ROS and MDA levels, but enhanced level of GSH. These findings unveiled that Fer-1 could reverse the ACR-induced ferroptosis in DRG neurons, which suggested the potential therapeutic application of Fer-1 in ACR-induced neurotoxicity. Fer-1 inhibited ferroptosis induced by ACR in DRG neurons. (a) The relative iron levels of DRG neurons were determined with ACR alone or in combination (ACR+ Fer-1) for 48 h (n = 3). (b–d) The concentrations of glutathione (b), malondialdehyde (c) and reactive oxygen species (d) of different groups were detected through ELISA assay kit (n = 3). *p < 0.05, **p < 0.01 in Tukey’s post-hoc comparisons test (A, B, C, D). Data are presented as mean ± SD. Abbreviations: ACR–acrylamide; DRG–dorsal root ganglia.
Discussion
Seeking a method to effectively repair injured neurons is an urgent challenge for the ACR-induced neural damage. Ferroptosis is a novel discovered form of programmed cell death with characterizations of iron and ROS accumulation. 29 Emerging studies unveiled that ferroptosis regulated many neurological disorders through different ferroptosis-related pathways.30,31 Nevertheless, the role of ferroptosis on the neurotoxicity caused by ACR still remains unknown. In this study, it was found the ACR caused neurotoxicity through triggering ferroptosis mainly in DRG neurons rather than Schwann cells. Moreover, we used Fer-1, a specific inhibitor of ferroptosis, to determine that ferroptosis modulation could provide a potential way to repair damaged nerve. These results contributed to understanding the mechanism underlying ACR-induced neurotoxicity and provided new avenues for future therapy development.
Interfering with the level of ferroptosis is a potential approach for the treatment of a variety of diseases. 32 Recent studies reported intracerebral hemorrhage-induced hemoglobin degradation triggered ferroptosis and caused irreversible neurons damage. Pyridoxal isonicotinoyl hydrazine (PIH), an iron chelator agent, could effectively inhibited the iron content and lipid peroxides and ultimately promoted the recovery of neurological functions. 33 FIN56, a specific ferroptosis inducer, suppressed cancer progression and triggered lysosomal membrane permeabilization in Glioblastoma through triggering ferroptosis in a TFEB-dependent manner. 34 Furthermore, Liu et al. revealed that GPX4 pathway promoted acquired chemo-resistance through inhibiting ferroptosis in lung cancer-derived brain metastasis. The use of GPX4 inhibitor, showed a promoting role of anti-cancer during cisplatin chemotherapy in vitro and in vivo, presenting a new strategy to overcome chemo-resistance in lung cancer with brain metastasis. 35
It was determined that the expression levels of neurotrophic factors including NGF, BDNF, VEGF and GDNF facilitated to a certain extent after ACR treatment, which indicated that the ACR-induced damage triggered the self-protective mechanism. However, the inhibition of cell proliferation and migration indicated that the neuronal self-protection was not enough to counteract the ACR-induced injury. Previous report had revealed a positive correlation between the concentration of ACR and the expression of BDNF in NB-1 cells. 36 Although we have investigated the ferroptosis modulation in ACR-caused neurotoxic effect on DRG neurons, the specific molecular mechanism remains to be further illustrated. Besides, in vivo experiments have not been carried out in this study, and animal experiments are warranted for further verification. Moreover, in our future work, the ferrostatin-1 experiments will be further performed on Schwan cells as a negative control to enrich our findings.
In conclusion, it was found that ACR inhibited the cell proliferation, migration and induced ferroptosis in DRG neurons. Furthermore, it was demonstrated that Fer-1 reversed ACR-induced the biological functions and promoted injured DRG neurons repair by suppressing ferroptosis. The current study revealed that Fer-1 is a promising molecular for ACR-caused neurotoxic effect and may contribute to the ferroptosis strategy for the treatment in the future.
Supplemental Material
Supplemental material - The effects of acrylamide-mediated dorsal root ganglion neurons injury on ferroptosis
Supplemental material for The effects of acrylamide-mediated dorsal root ganglion neurons injury on ferroptosis by Shuai An, Jingfei Shi, Zheng Li, Mingli Feng and Guanglei Cao in Human & Experimental Toxicology
Footnotes
Acknowledgements
The authors would like to thank the members of the department of Orthopedics at Xuanwu Hospital, Capital Medical University, for their assistance in data collection and surgery.
Authors’ contributions
S.A. conceived the experiment, S.A., J.S. and Z.L. conducted the experiment, M.F., G.C, S.A. analyzed the results. All authors reviewed the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The research was supported by the Capital Health Research and Development of Special (2020-4-2018).
Ethics approval
This study was approved by the Ethics Committee of Xuan Wu Hospital, Capital Medical University (Beijing, China) (Number [2019]175).
Data availability statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Supplemental Material
Supplemental material for this article is available online.
Appendix
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.