
Abstract
Introduction
Quercetin has been reported to inhibit the growth of oral squamous cell carcinoma (OSCC), but the mechanism remains unclear. Therefore, our study aimed to investigate the involvement of sirtuin 3 (SIRT3) and the autophagy-dependent form of cell death, ferroptosis, in the pathogenesis of OSCC, and observe the impacts of quercetin on ferroptosis and SIRT3/AMPK/mTOR-mediated autophagy.
Methods
SIRT3 knock out or overexpressing SCC15 cell line was generated, treated with indicated drugs, and malondialdehyde (MDA) and ROS levels were measured. Roles of SIRT3 in regulating autophagy-mediated ferroptosis were assessed by immunoprecipitation and Western blotting.
Results
SIRT3 overexpression increased levels of MDA and ROS, reducing cell viability, and SIRT3 knockout produced the opposing effect. SIRT3 overexpression upregulated ATG16L1 expression and the conversion of LC3-Ⅰ to LC3-Ⅱ, triggering autophagy. Suppression of autophagy by ATG16L1 knockout impaired SIRT3-triggered ferroptosis. Use of an AMPK inhibitor antagonized the induction of ferroptosis by SIRT3 in SCC15 cells, indicating the involvement of the AMPK/mTOR pathway. Additionally, quercetin significantly increased the levels of SIRT3, p-AMPK, ATG16L1, and the ratio of LC3-Ⅱ/Ⅰ, but reduced cell viability and p-mTOR in SCC15 cells. Autophagy and AMPK inhibitors, or SIRT3 deletion significantly antagonized the impacts of quercetin on the autophagy-mediated ferroptosis in cancer cells.
Discussion
SIRT3 overexpression activated the AMPK/mTOR pathway and triggered ATG16L1-mediated autophagy, promoting ferroptosis in SCC15 cells, and we proposed that quercetin may be a promising therapeutic drug for OSCC.
Introduction
Oral squamous cell carcinoma (OSCC) is the most common and aggressive oral malignancy, accounting for about 90% of oral cancers worldwide. 1 The disease is treated by surgical excision and chemoradiotherapy but prognoses remain unsatisfactory. 2 OSCC pathogenesis has been associated with cytopathological and genomic heterogeneity but molecular mechanisms have yet to be elucidated, although ferroptosis has been implicated.3,4
Autophagy is a form of programmed cell death which eliminates aggregates of misfolded protein and damaged organelles. 5 Excessive ROS production has been shown to trigger lipid peroxidation and decrease antioxidant enzyme activity to induce autophagy in OSCC. 4 By comparison, ferroptosis has been linked to excessive ROS accumulation, iron-dependent lipid peroxidation and redox dysfunction. 6 Interactions between autophagy and ferroptosis remain to be elucidated.
Sirtuin 3 (SIRT3) is a prototypical NAD+-dependent mitochondrial protein deacetylase, involved in the modulation of oxidative stress, 7 and has been linked to the pathogenesis of various cancers, including OCSS. SIRT3 has been identified as an OSCC tumor suppressor and to regulate mitochondrial function, including ATP-production, nutrient oxidation, ROS levels and cell death.8,9 SIRT3 and AMP-activated protein kinase (AMPK) interact to modulate the mitochondrial antioxidant network and SIRT3 activation of AMPK has been implicated in autophagy activation. 10 SIRT3/AMPK/autophagy signal axis has been reported to involve in the invasion and metastasis of breast cancer cells and cervical cancer cells.11,12 AMPK is thought to act through mTOR and SIRT3 to stimulate ferroptosis in tumors and, therefore, the impact of SIRT3 on cell viability, autophagy and ferroptosis in OSCC may involve the AMPK/mTOR cascade. As part of our study, we further explored how quercetin affects OSCC cells. Quercetin, derived from flavonols, also has an anti-cancer role in multiple cancer cells. 13 We investigated how quercetin triggers ferroptosis in SCC15 cells via regulating SIRT3/AMPK/mTOR-dependent autophagy.
Materials and methods
Cell culture and drug treatment
SCC15 cell line was purchased from the Chinese Academy of Sciences Cell Bank (Shanghai, China). Cells (1 × 106 cells/mL) were cultured in DMEM supplemented with 10% FBS at 37°C, 95% humidity and 5% CO2. When cells reached 80% confluence, cells were digested by trypsin after removing the medium, and then were sub-cultured. In addition, when 70% confluence was reached and treated with drugs at for 24h. Quercetin (#Q4951, Sigma-Aldrich, ≥95% (HPLC)), erastin (#E7781, Sigma-Aldrich, ≥98% (HPLC), powder, ferroptosis inducer), sorafenib (#Y0002098, Sigma-Aldrich, ≥98% (HPLC), powder, ferroptosis inducer), RSL3 (#SML2234, Sigma-Aldrich, ≥98% (HPLC), powder, ferroptosis inhibitor), liproxstatin-1 (#SML1414, Sigma-Aldrich, ≥98% (HPLC), powder, ferroptosis inhibitor), ZVAD-FMK (#V166, Sigma-Aldrich, ≥98% (TLC), solid, apoptosis inhibitor), necrostatin-1 (#N9037, Sigma-Aldrich, ≥98% (HPLC), powder, necroptosis inhibitor), 3-MA (#189490, Sigma-Aldrich, ≥95% (HPLC), powder, autophagy inhibitor), and BML-275 (#P5499, Sigma-Aldrich, ≥98% (HPLC), powder, AMPK inhibitor) were dissolved in DMSO and stored at −20°C. Control cells were treated with an equal volume of DMSO.
Plasmid transfection
Gene knockout was achieved by Lipofectamine 2000 (Thermo Fisher Scientific) transfection for 48h, according to the manufacturer’s instructions, with shRNA duplexes complementary to the SIRT3 or ATG16L1 genes or negative control shRNAs, all synthesized by GenePharma (Shanghai, China). SIRT3 upregulation was achieved by transfection with pcDNA3.1-SIRT3 or empty vector (control), synthesized by GenePharma.
Cell viability assay
SCC15 cells were grown in 96-well plates, medium removed and 100 μL fresh medium containing 10% CCK-8 (cat#BN15201-500T; Biorigin) added with incubation for 2 h at 37°C. The optical densities were recorded at 450 nm by microplate reader (Bio‐Rad, Hercules, CA).
Lipid peroxidation assay
Malondialdehyde (MDA) was determined in cell lysates by Lipid Peroxidation Assay Kit (ab233471, Abcam, CA, USA), according to the manufacturer’s directions.
Measurement of ROS levels
Intracellular ROS were detected by an oxidation sensitive fluorescent probe, DCFH-DA (ab113851, Abcam, CA, USA), according to the manufacturer’s directions. Briefly, 20 μM DCFH-DA reagent was used to stain SCC15 cells for 30 min at 37°C in the dark and fluorescence measured at 535 nm by flow cytometer (Leica, Germany).
Detection of SIRT3 mRNA
Total RNA was isolated using TRIzol reagent (Invitrogen, USA), according to the manufacturer’s directions, reverse-transcribed with PrimeScript™ RT reagent kit (Takara, Japan) and qPCR performed using QuantiTect SYBR Green PCR Kit (Takara, Japan). Relative SIRT3 mRNA expression was normalized by β-actin and calculated using the 2−ΔΔCT method. The primers were as follows: SIRT3, forward 5′- ACCCAGTGGCATTCCAGAC-3′, reverse 5′- GGCTTGGGGTTGTGAAAGAAG-3′; β-actin, forward 5′-AGCCTTCCTTCTTGGGTATGGAATC-3′, reverse 5′-GGAGCAATGATCTT GATCTTCATGG-3′.
Western blotting and immunoprecipitation
For Western blotting: cells were rinsed with PBS, total protein extracted using RIPA buffer (Thermo Scientific, USA) on ice for 30 min. And protein concentrations measured by BCA Protein Assay Kit (Beyotime, China). 40 μg total protein was separated with 10%–12% SDS-PAGE, transferred to PVDF membrane (Merck Millipore, USA) and blocked with 5% skimmed milk for 1h. Membranes were incubated with primary antibodies: anti-ATG3 (ab108251, Abcam), anti-ATG4A (ab108322), anti-BECN1 (ab207612), anti-ATG7 (ab52472), anti-ATG9A (ab108338), anti-ATG16L1 (ab187671), anti-ATG12 (ab303488), anti-ATG5 (ab228668), anti-LC3B (ab48394), anti-SIRT3 (ab217319), anti-AMPK (ab32518), anti-p-AMPK (133448), anti-mTOR (ab134903), anti-p-mTOR (ab109268), and anti-β-actin (ab7817) overnight at 4°C. All primary antibodies were diluted for 1:1000. HRP-conjugated secondary antibodies (dilution 1:2000) were added for 1 h at room temperature and lanes visualized with chemiluminescence (Bio-Rad, CA, USA).
Immunoprecipitation: protein extracts were incubated with antibodies overnight at 4°C, protein A/G plus agarose beads (Santa Cruz Biotechnology, USA) added for 2h at 4°C, rinsed 5 times with lysis buffer and Western blotting performed.
Statistical analysis
All data are presented as the mean ± SD. Analysis was performed using GraphPad Prism software with comparisons between 2 groups by unpaired two-tailed Student'st test. A value of p < .05 was considered statistically significant.
Results
SIRT3 expression increased during ferroptosis in SCC15 cells
Erastin, sorafenib and RSL3-mediated reduction of SCC15 cell viability was antagonized by the ferroptosis inhibitor, liproxstatin-1, but not by the apoptosis inhibitor, ZVAD-FMK, or necroptosis inhibitor, necrostatin-1 (Figure 1(a)). Erastin, sorafenib and RSL3 increased levels of MDA and ROS, indicating increased lipid peroxidation, and these effects were blocked by liproxstatin-1 (Figure 1(b), and (c)). The expression levels of SIRT3 mRNA in erastin-, sorafenib-, and RSL3-treated cells were 2.36, 2.51, and 2.81 times higher than control cells, respectively (Figure 1(d)). Moreover, erastin, sorafenib and RSL3 treatment did not affect the expressions of apoptosis (caspase-3 and caspase-9), programmed necrosis biomarkers (CypD), cell apoptotic rate, but promoted the reduction of mitochondrial membrane potential (Supplemental Figure 1). Erastin, sorafenib and RSL3 increased SIRT3 protein in SCC15 cells, indicating the involvement of SIRT3 in ferroptosis (Figure 1(e), and (f)). SIRT3 expression increased during ferroptosis in SCC15 cells. SCC15 cell line was exposed to 10 μM erastin, 10 μM sorafenib or 2.5 μM RSL3 in the presence or absence of 100 nM liproxstatin-1, 10 μM ZVAD-FMK or 10 μM necrostatin-1for 24 h. (a) Cell viability; (b) MDA levels; (c) ROS levels in SCC15 cells. (d) SIRT3 mRNA measured by qRT-PCR. (e), (f) SIRT3 protein detected by Western blotting. *p < .05 compared with control group.
SIRT3 regulated ferroptosis in SCC15 cells
Viability and ferroptosis in SIRT3-overexpressing SCC15 cells were evaluated (Figure 2(a)). SIRT3 overexpression reduced the cell viability (Figure 2(b)) and increased ferroptosis-associated events, including increased MDA (Figure 2(c)) and ROS were seen (Figure 2(d)). SIRT3 was also downregulated in SCC15 cells (Figure 2(e)) and the effects of treatment with RSL3, which reduced cell viability and increased levels of MDA and ROS, were antagonized by SIRT3 knock out (Figure 2(f)–(h)). SIRT3 knockout appeared to confer resistance to ferroptosis in OSCC cells following RSL3 treatment. SIRT3 regulates ferroptosis in OSCC cells. SCC15 cells overexpressing SIRT3 were constructed. (a) SIRT3 protein was measured using Western blotting. (b) Cell viability; (c) MDA; (d) ROS. SIRT3 knockout SCC15 cells were exposed to 2.5 μM RSL3 for 24 h. (e) SIRT3 protein was measured using Western blotting. (f) Cell viability; (g) MDA; (h) ROS. *p < .05 compared with control group; #p < .05 compared with sh-RNA + RSL3 group.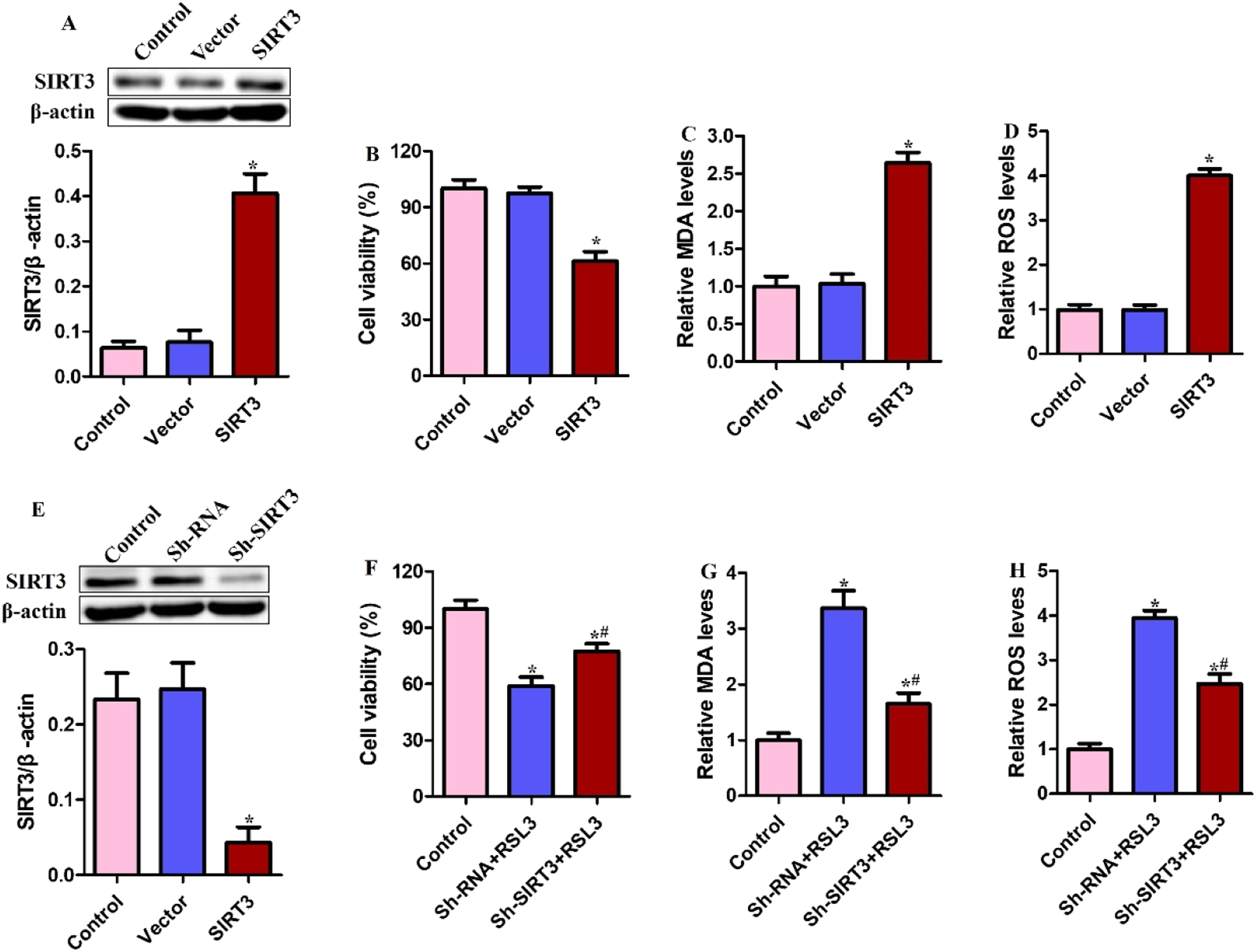
Ferroptosis induced by SIRT3 was associated with activation of autophagy
Autophagy-associated proteins were investigated and SIRT3 overexpression induced ATG16L1 expression but not that of other autophagy-associated proteins, such as ATG4A, ATG12-ATG5, BECN1, ATG7 and ATG9A (Figure 3(a)). Activity of the conserved ATG12-ATG5-ATG16L1 complex is known to be involved in the conversion of LC3-Ⅰ to LC3-Ⅱ and autophagosome formation.
14
SIRT3 overexpression induced formation of the ATG12-ATG5-ATG16L1 complex in SCC15 cells (Figure 3(b)) and triggered the conversion of LC3-Ⅰ to LC3-Ⅱ (Figure 3(c)). The induction of ferroptosis by SIRT3 overexpression was linked to activation of autophagy. SCC15 cells overexpressing SIRT3 were constructed. (a) ATG3, ATG4A, ATG12-ATG5, BECN1, ATG7, ATG9A and ATG16L1 protein were detected. (b) The ATG12-ATG5-ATG16L1 protein complex was determined by immuno-coprecipitation. (c) LC3-Ⅰ and LC3-Ⅱ protein were measured. SCC15 cells overexpressing SIRT3 or with ATG16L1 knockout were exposed to 2.5 μM RSL3 for 24 h. (d) ATG16L1 protein was detected; (e) Cell viability; (f) MDA; (g) ROS. *p < .05 compared with control or vector + RSL3 group; #p < .05 compared with SIRT3 + RSL3 group.
Cells transfected with the sh-ATG16L1 plasmid were used to stimulate autophagy inactivation and perform reverse validation of the stimulation of ferroptosis by SIRT3. ATG16L1 knock out reduced cell viability and increased levels of MDA and ROS in RSL3-treated cells and these effects were antagonized by SIRT3 overexpression (Figure 3(d)–(g)). The sh-ATG16L1 plasmid appeared to reduce SIRT3-triggered ferroptosis in SCC15 cells.
SIRT3 overexpression induced autophagy activation and ferroptosis by activating the AMPK/mTOR pathway
AMPK has been reported to modulate mTOR signaling and positively regulate autophagy. Phosphorylation states of AMPK and mTOR were assessed after SIRT3 overexpression in SCC15 cells. SIRT3 overexpression was found to be involved in AMPK activation, evidenced by increased p-AMPK and reduced p-mTOR (Figure 4(a)). SIRT3 overexpression triggered ATG16L1 expression and the conversion of LC3-Ⅰ to LC3-Ⅱ but these effects were antagonized by the AMPK inhibitor, BML-275 (Figure 4(b)). These findings were mirrored by those regarding ferroptotic events, including cell viability, MDA and ROS in SCC15 cells (Figure 4(c)–(e)). SIRT3 overexpression appeared to induce autophagy and ferroptosis by activating the AMPK/mTOR pathway. Inhibition of AMPK blocked SIRT3-triggered ferroptosis. SCC15 cells overexpressing SIRT3 were exposed to 2.0 μM BML-275 for 24 h. (a) AMPK, p-AMPK, mTOR and p-mTOR determined by Western blotting. (b) ATG16L1, LC3-Ⅰ and LC3-Ⅱ protein were measured. (c) Cell viability; (d) MDA; (e) ROS. * <0.05 compared with control group; #p < .05 compared with SIRT3 group.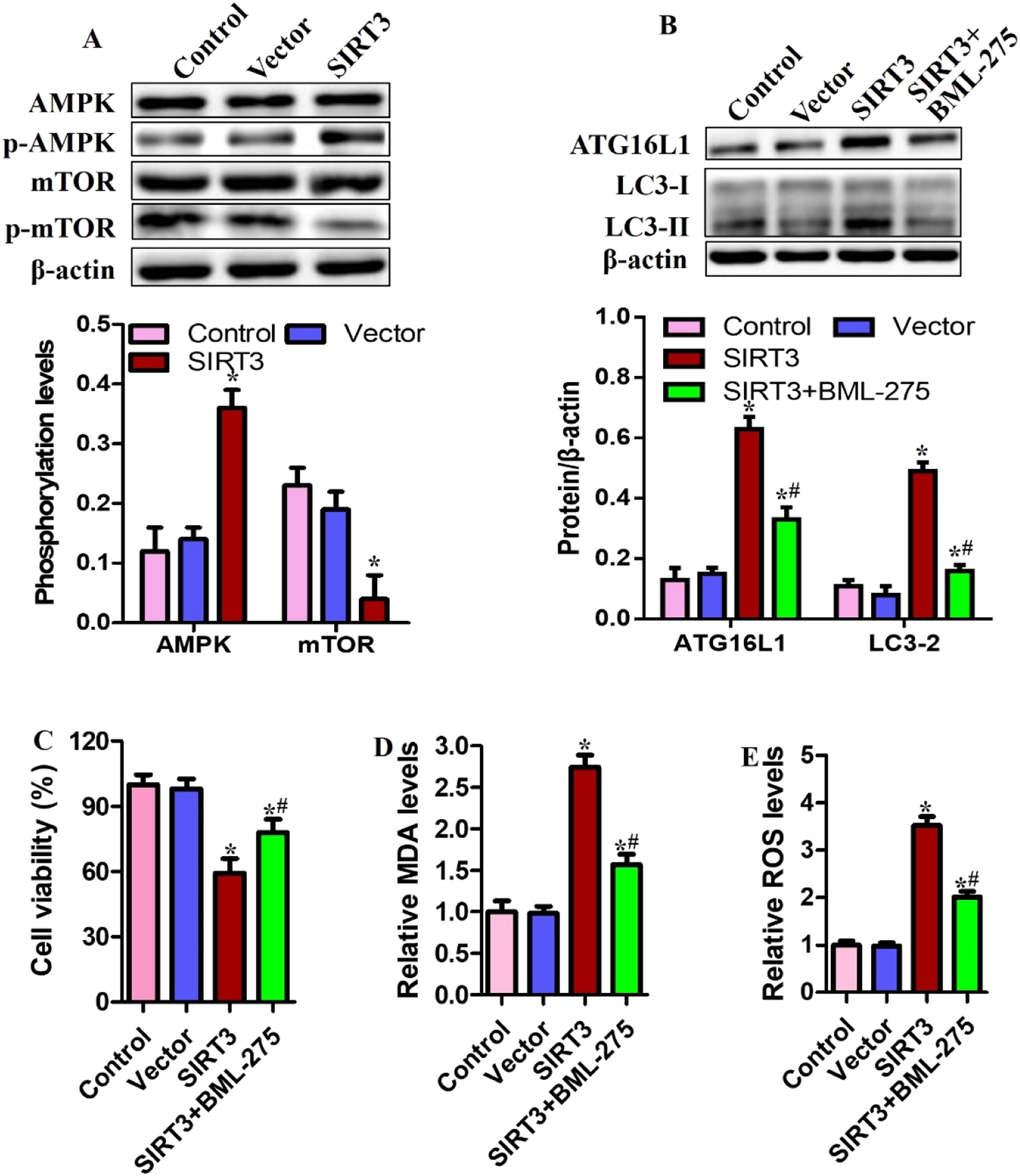
Quercetin induces ferroptosis through the SIRT3/AMPK/mTOR-triggered autophagy activation
This study further explored the effects of the SIRT3/AMPK/mTOR-mediated autophagy in quercetin-triggered ferroptosis in SCC15 cell line. As shown in Figure 5(a), quercetin significantly decreased the viability of SCC15 cells in concentration- and time-dependent manners. Quercetin also significantly induced the levels of MDA and ROS at a concentration-dependent way (Figure 5(b), and (c)). Moreover, quercetin significantly induced the expression levels of SIRT3, p-AMPK, ATG16L1 and the ratio of LC3-Ⅱ/Ⅰ but reduced the expression of p-mTOR (Figure 5(d), and (e)). In addition, the autophagy inhibitor (3-MA) exposure significantly weakened the impacts of quercetin on cell viability and the levels of MDA and ROS (Figure 5(f)–(h)). Quercetin induces ferroptosis through the SIRT3/AMPK/mTOR-triggered autophagy activation. SCC15 cell line was exposed to different doses of quercetin (0-160 μM) for 12, 24, and 48 h, respectively; (a) Cell viability was detected by CCK-8 assay. SCC15 cell line was treated with different concentrations of quercetin (0, 20, 40, and 80 μM) for 24 h; (b) MDA, (c) ROS were determined; (d, e) the expressions of SIRT3, AMPK, p-AMPK, mTOR, p-mTOR, ATG16L1, and LC3-Ⅱ/Ⅰ were also determined by Western blotting. SCC15 cell line was co-treated with 3-MA (2.0 μM) and quercetin (40 μM) for 24 h, (f) cell viability, (g) MDA, (h) ROS were also measured. *p < .05, compared with control group; #p < .05, compared with quercetin group.
Our study further verified whether quercetin induced ferroptosis in SCC15 cells by activating SIRT3/AMPK/mTOR axis. SIRT3 knockout significantly decreased the expression levels of p-AMPK, ATG16L1 and the ratio of LC3-Ⅱ/Ⅰ but induced the expression of p-mTOR in quercetin-treated cells (Figure 6(a), and (b)). Similarly, SIRT3 deletion increased cell viability, whereas significantly reduced the levels of MDA and ROS in quercetin-treated cells (Figure 6(c)–(e)). Moreover, BML-275 treatment also significantly induced cell viability and decreased the levels of MDA and ROS in quercetin-treated cells (Figure 6(f)–(h)). Our findings indicated that quercetin induced the ferroptosis in SCC15 cells via promoting SIRT3/AMPK/mTOR-mediated autophagy activation. Inhibition of SIRT3 or AMPK decreased ferroptosis sensitization in quercetin -exposed cells. SCC15 cells low-expressing SIRT3 were exposed to 40 μM quercetin for 24 h; (a), (b) Effects of SIRT3 knockout on autophagy-related proteins in quercetin-treated cells. (c)-(e) Effects of SIRT3 knockout on cell viability, MDA, and ROS levels in quercetin-treated cells. SCC15 cell line was co-treated with BML-275 (2.0 μM) and quercetin (40 μM) for 24 h, (f)-(h) Impacts of BML-275 on cell viability, MDA, and ROS levels in quercetin-treated cells. *p < .05, compared with untreated cells; #p < .05, compared with quercetin group.
Discussion
SIRT3 was shown to increase during autophagy-dependent ferroptosis in OSCC cells. SIRT3 overexpression promoted sensitivity to ferroptosis by activating AMPK/mTOR-mediated autophagy.
Ferroptosis is known to be stimulated by excessive accumulation of cellular iron and ROS and ferroptosis regulatory factors have been shown to mediate the progression of OSCC and anti-cancer responses with implications for prognostic evaluation and treatment.15–17 Some researchers showed that a curcumin analog inhibits growth of temozolomide-resistant OSCC via ferroptosis stimulation and pembrolizumab initiated ferroptosis and suppressed growth of OSCC cells.18,19 The current findings confirm that the inducers of ferroptosis, erastin, sorafenib and RSL3, inhibited SCC15 cell viability (Figure 1) but many questions remain to be answered. Individual ferroptosis inducers differ by cell selectivity and this must be taken into account during the search for new OSCC drug targets.
The current study verified the regulatory role of SIRT3 in ferroptosis in OSCC. Overexpression of SIRT3 is known to regulate mitochondrial function and ROS levels and to promote the protein acetylation which underlies cellular resistance to ferroptotic events.20,21 SIRT3 expression was found to increase in the OCSS cell line of the current study after treatment with ferroptosis inducers. SIRT3 overexpression reduced cell viability and triggered ferroptosis in RSL3-treated cells whereas SIRT3 depletion had the opposing effect (Figure 2). Thus, the participation of SIRT3 in ferroptosis in OCSS cell line was confirmed.
Autophagy and ferroptosis are known to cross talk through complex feedback loops. 22 Ferroptosis can lead to the excess accumulation of lipid peroxidation products, which are participant in the induction of autophagosome formation. Autophagy activation also seems to be a key driver of ferroptosis. Moreover, certain selective types of autophagy, such as ferritinophagy, lipophagy, clockophagy, and chaperone-mediated autophagy, trigger ferroptotic events of OSCC cells the degradation of ferroptosis repressors, leading to iron and lipid metabolism. 22 Ferroptosis is associated with autophagosome formation, consistent with the notion that both lipid peroxidation and oxidized lipids activate autophagy. During autophagy, autophagosomes engulf cytoplasmic components. At the same time, LC3-I is conjugated to phosphatidylethanolamine to form LC3-II, which is recruited to autophagosomal membranes. Therefore, lysosomal turnover of the autophagosomal marker LC3-II reflects autophagic activity, and the ratio of LC3-II to LC3-I has become a reliable marker for monitoring autophagy and autophagy-related processes. 22 SIRT3 overexpression increased ATG16L1 expression and the conversion of LC3-Ⅰ to LC3-Ⅱ in the current study (Figure 3), suggesting a role in autophagy activation. SIRT3 has previously been reported to activate autophagy and prevent doxorubicin-induced senescence in lung cancer cells 23 and excessive activation of autophagy stimulated induction of ferroptosis in cancer cells.24,25 However, the functional contributions of SIRT3 to autophagy and ferroptosis remain poorly understood. The involvement of ATG16L1 in autophagy has been reported and the protein was found to interact with the ATG12-ATG5 complex to increase LC3 lipidation.26,27 Indeed, ATG16L1 is known to be involved in the autophagic degradation of aggregates of misfolded protein and damaged organelles.5,28 SIRT3 overexpression induced ATG16L1 upregulation and stimulated ferroptosis in the OSCC cells of the current study. ATG16L1 depletion partially reversed SIRT3-triggered autophagy and ferroptosis.
SIRT3 has been shown to cause AMPK phosphorylation, reducing mTOR activity and activating autophagy. 21 AMPK is known to modulate autophagy via ATG16L1 and the AMPK/mTOR cascade was likely to be involved. 29 SIRT3 overexpression increased the phosphorylation state of AMPK and reduced mTOR activity during the present study and the AMPK inhibitor, BML-275, blocked the SIRT3 effects on autophagy and ferroptosis (Figure 4). These results are consistent with those of a previous investigation. 30 We conclude that SIRT3 triggers ATG16L1-mediated autophagy by activating the AMPK/mTOR cascade.
As a bioactive compound of herbal origin, quercetin was found to have many pharmacological effects. 13 It has reported that quercetin has an anti-cancer effect via multiple signaling pathways, including cell survival, apoptosis, and necroptosis. 31 Quercetin regulates autophagy in mitochondria through the SIRT1/SIRT3-FOXO3a axis. 32 Quercetin also can induce ferroptosis in cancer cells by multiple mechanisms. 33 Moreover, it has been demonstrated that quercetin inhibits cell growth and promotes apoptosis in OSCC cells. 34 Thus, the present study expands the list by including autophagy and ferroptosis in OSCC. Our study found that quercetin can promote ferroptosis through SIRT3-dependent autophagy in SCC15 cells, confirming that quercetin exerts these effects partially by activating the AMPK/mTOR cascade (Figures 5 and 6). Therefore, quercetin may serve as an adjuvant therapy for chemotherapeutic drugs in OSCC. Moreover, the main limitation in our study was the absence of in vivo studies, and we will perform these investigations to prove our in vitro conclusions.
Conclusion
In summary, SIRT3 was found to be a promising positive regulator of ferroptosis in OSCC and regulated ATG16L1-mediated autophagy and ferroptosis via upregulation of the AMPK/mTOR signaling pathway in OSCC cells. Considering the activation of the SIRT3/AMPK/mTOR axis in OSCC, a ferroptosis agonist, quercetin may be a promising treatment agent.
Supplemental Material
Supplemental Material - Activation of SIRT3/AMPK/mTOR-mediated autophagy promotes quercetin-induced ferroptosis in oral squamous cell carcinoma
Supplemental Material for Activation of SIRT3/AMPK/mTOR-mediated autophagy promotes quercetin-induced ferroptosis in oral squamous cell carcinoma by Jin Wang, Jia-hui Yang, Di Xiong, and Ling Chen in Human & Experimental Toxicology.
Footnotes
Acknowledgments
Author contributions
JW and JHY conceived and designed the experiments, DX analyzed and interpreted the results of the experiments, LC performed the experiments.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Health and Family Planning Commission of Wuhan Scientific Research Project (WZ22Q12) and the Shizhen Talent Program of Hubei Province for Scientific Research ([2024] No. 256). The authors declare that all research was conducted with full compliance to ethical standards and institutional guidelines.
Ethical statement
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.