
Abstract
Cigarette smoke (CS)-caused ferroptosis was involved in the pathogenesis of COPD, but the role of ferroptosis in lung epithelial injury and inflammation is not clear. Rats were treated with CS or CUR and BEAS-2B cells were exposed to CS extract (CSE), ferrostatin-1 (Fer-1), deferoxamine (DFO), or CUR to detect reactive oxygen species (ROS) accumulation, lipid peroxidation, iron overload, and ferroptosis-related protein, which were the characteristic changes of ferroptosis. Compared with the control group, CSE-treated BEAS-2B cells had more cell death, higher cytotoxicity, and lower cell viability. The infiltration of inflammatory cell around the bronchi in the CS group of rats was more than that in the normal group. Meanwhile, CSE/CS elevated the levels of interleukin-6 and tumor necrosis factor-α in BEAS-2B cells and bronchoalveolar lavage fluid of rats. Besides, accumulative ROS and depleted glutathione was observed in vitro. In BEAS-2B cells and lung tissues of rats, CSE/CS increased malondialdehyde and iron; down-regulated solute carrier family 7, glutathione peroxidase 4, and ferritin heavy chain levels; and up-regulated transferrin receptor level. These changes were rescued by pretreatment of Fer-1 or DFO in vitro, and mitigated by CUR in vitro and in vivo. Collectively, this study reveals that ferroptosis was involved in lung epithelial cell injury and inflammation induced by CS, and CUR may alleviate CS-induced injury, inflammation, and ferroptosis of lung epithelial cell.
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic airway inflammatory disease characterized by progressive expiratory airflow limitation. COPD has been the fourth leading cause of mortality in the world; however, current treatment for COPD is poorly effective, and the bottleneck of treatment has not been broken. 1 COPD is mainly related to cigarette smoke (CS) exposure, which could cause aberrant airway inflammation and recurrent oxidative stress via apoptosis and necroptosis, leading to extensive lung injury and disease aggravation. 2 Yoshida et al. 3 found that CS-caused lung epithelial cell ferroptosis was involved in the pathogenesis of COPD. However, it is not clear how CS induces lung injury, inflammation, and ferroptosis, and whether lung injury and inflammation are related to ferroptosis.
Ferroptosis is a novel form of cell death, which is different from necrosis, apoptosis, and autophagy in heredity, biochemistry, and morphology. Ferroptosis is characterized by impaired repair capacity of lipid peroxide, accumulation of lipid-reactive oxygen species (ROS), glutathione (GSH) depletion, and intracellular iron overload. 4 During ferroptosis, the destruction of iron metabolism regulatory systems, such as ferritin heavy chain (FTH1) and transferrin receptor (TFR1), leads to labile iron accumulation, which evokes the Fenton reaction and further induces ROS-caused phospholipid peroxidation. 5 Increasing studies have suggested that ferroptosis is closely associated with many disease processes, such as lung, 3 renal, 6 and brain injuries. 7 Therefore, the inhibition of ferroptosis is expected to provide potential therapeutic strategies in many diseases.
Curcumin (CUR) is a yellow polyphenol compound derived from plant turmeric, which has shown anti-inflammatory, antioxidant, and anticancer effects. 8 CUR may play a crucial role in the treatment of COPD, and this role may be decided by the strong antioxidant capacity of CUR related to scavenging excessive ROS. 9 In animal models of COPD, CUR down-regulated the inflammatory cytokines interleukin-6 (IL-1β), IL-6, and tumor necrosis factor-α (TNF-α) and suppressed neutrophil recruitment in bronchoalveolar lavage fluid (BALF). 10 Emerging research showed that CUR could reduce renal damage by decreasing ferroptotic cell death. 11 However, the specific role of CUR in CS-caused ferroptosis and the intrinsic mechanism remain unclear.
In this study, whether CS exposure could induce epithelial cell injury, inflammation, and ferroptosis was verified, and the role of ferroptosis in the epithelial cell injury and inflammation induced by CS was explored in vitro and in vivo. Then, whether the injury, inflammation, and ferroptosis of lung epithelial cell were alleviated by CUR was further investigated. The findings in this study are expected to provide a novel therapeutic target for patients with COPD.
Methods
Reagents and antibodies
CUR (C1386), ferrostatin-1 (Fer-1, SML0583), and deferoxamine (DFO, D9533) were purchased from Sigma-Aldrich. Cell counting kit-8 (CCK-8) assay kit (C6005) was purchased from US Everbright Inc. The detection kits for lactate dehydrogenase (LDH, A020-2), ROS (E004-1), malondialdehyde (MDA, A003-1), and tissue iron (A039-2) were purchased from Nanjing Jiancheng Bioengineering Institute. GSH assay kit (S0053) was purchased from Beyotime Biotechnology. Intracellular iron colorimetric assay kit (E1042) was purchased from Applygen Technologies Inc. Calcein-AM/PI double stain kit (40747ES76) was purchased from YEASEN Biotechnology. The human IL-6 enzyme linked immunosorbent assay (ELISA) kit (EK0410), rat IL-6 ELISA kit (EK0412), human TNF-α ELISA kit (EK0525), and rat TNF-α ELISA kit (EK0526) were purchased from BOSTER Biotechnology. The glutathione peroxidase 4 (GPX4) (ab125066) antibody was purchased from Abcam. The antibodies solute carrier family 7 member (SLC7A11, A2413), FTH1 (A1144), TFR1 (A5865), and β-actin (AC004) were purchased from ABclonal.
Preparation of cigarette smoke extract
CSE was extracted from Daqianmen cigarettes (tar: 10 mg, nicotine: 1 mg, carbon monoxide: 12 mg; Tianjin, China) as previously reported with minor modifications. 3 In brief, five cigarettes were burned in turn, and the CS was sucked into the syringe and bubbled into 10 mL of sterile phosphate buffered saline (PBS) in bubble absorption bottle. The CSE solution was filtered (0.22 μm; Millipore, USA) to remove the bacteria and insoluble particles. CSE preparations were designated as a 100% CSE solution.
Cell culture
Human bronchial epithelial cell line BEAS-2B was kindly provided by the Laboratory of Lung Development and Diseases at Nankai University (Tianjin, China). The BEAS-2B cells were cultured in Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS). The cells were incubated under humidified conditions with 5% CO2 at 37°C. BEAS-2B cells were treated with 5 μM Fer-1 or 80 μM DFO 1 h before 2.5% CSE treatment for 24 h.
Cell death assay
In accordance with the manufacturer’s protocol, the LDH assay kit, and CCK-8 assay kit were used to determine cytotoxicity and cell viability, respectively, to analyze cell death. The BEAS-2B cells were seeded in a 96-well microplate with 5000 cells/well. After 24 h, the cells were incubated in non-FBS DMEM containing CSE (0, 1, 2.5, and 5%) for 24 h. Cell supernatant was collected for the LDH assay. Then, cells were washed with non-FBS DMEM twice, new non-FBS DMEM at 100 μl/well and CCK solution at 10 μl/well were added and incubated for an additional 2 h. The absorbance at 450 nm was detected by a microplate reader (BioTek, Vermont, USA). The calculation methods are as following: cytotoxicity (%) = (Atreatment − Acontrol)/(Amax − Acontrol) × 100, cell viability (%) = (Atreatment − Ablank)/(Acontrol − Ablank) × 100.
Animal models
Sprague-Dawley rats (male, seven weeks old, and 150 ± 20 g) were provided by SiPeiFu Biotechnology Company (Beijing, China). They were divided randomly into four groups of five as follows: normal control (NC), CS, CUR, and CS + CUR group. The rats were housed in a pathogen-free room with a light-dark cycle of 10:14 h, a temperature of 26 ∼ 28°C, and a relative humidity of 40% ∼ 60%. Except during the CS exposure period, rats are free to eat and drink water. After the rats were raised for a week, those in the CS and CS + CUR groups were passively exposed to CS by using a homemade smoke exposure device as described previously. 12 The rats were exposed to 15 cigarettes for 30 min each time, twice a day (at 9 a.m. and 9 p.m), and 6 days a week for 16 weeks. And the volume fraction of CS in the smoke exposure device was about 15% (V/V). The rats in the NC and CUR groups are exposed to normal air. The rats in the CUR and CS + CUR groups were intragastrically administered with CUR (100 mg/kg/day), while those in the NC and CS groups were given sodium carboxymethyl cellulose at 8 a.m. every day for 16 weeks. All experimental procedures were performed in accordance with the guidelines issued by the Guide for the Care and Use of Laboratory Animals of Tianjin Medical University.
Histologic analysis
The rats were anesthetized by intraperitoneal injection of 10% chloral hydrate (0.3 mL/100g) and sacrificed by cardiac puncture and bloodletting. BALF was collected by repeatedly lavaging the lungs with PBS through a tracheostomy cannula. The right lung was resected and frozen with liquid nitrogen for western blot and other experiments, and the left lung was filled with 10% neutral formalin, resected, fixed, paraffin-embedded, and cut into 4 μm sections. Then, the dewaxed sections were stained by hematoxylin and eosin to evaluate lung epithelial injury.
Levels of inflammatory cytokines
The concentrations of IL-6 and TNF-α in supernatants of BEAS-2B cells and BALF of rats were measured using the corresponding ELISA kit in accordance with the manufacturers’ protocol.
Western blot analysis
Total proteins were isolated from BEAS-2B cells and lung tissue of rats by using RIPA buffer and the concentration was measured using a BCA protein kit. The same amount of denatured protein (cells: 30 μg; lung tissues: 50 μg) of each sample was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. They were transferred onto polyvinylidene fluoride membranes, and the membranes were incubated with primary antibodies at 4°C overnight. Then, the membrane was incubated with goat anti-rabbit/mouse IgG and HRP-linked antibodies as a secondary antibody for 2 h at room temperature. The protein bands were detected by electrochemical luminescence, and the band density was analyzed using the Tanon GIS digital image analysis system (Tanon, Shanghai, China). The relative protein levels were normalized to β-actin.
Determination of reactive oxygen species generation
Intracellular ROS levels were detected with peroxide-sensitive fluorescent probe 2′7′-dichlorofluorescin diacetate (DCFH-DA) in accordance with the manufacturer’s instructions. In brief, DCFH-DA was diluted to 10 μM in serum-free medium. The BEAS-2B cells were incubated with diluted DCFH-DA at 37°C for 1 h. Fluorescence intensity was measured via flow cytometry in accordance with the FITC fluorescence detection conditions.
Malondialdehyde, glutathione, and iron assay
BEAS-2B cells were washed with PBS, harvested, and then centrifuged to collect cell precipitates. The lung tissue was frozen in liquid nitrogen and then ground into powder. According to the manufacturer’s instructions, the contents of MDA, GSH, and iron in BEAS-2B cells and lung tissues of rats were measured with corresponding kit.
Statistical analysis
Data were shown as mean ± standard deviation. One-way ANOVA was used for comparison among multiple groups, and LSD was used to compare the differences between two groups. p < 0.05 was considered statistically different. Statistical analysis was performed using GraphPad Prism 7.0 software.
Results
Cigarette smoke extract induces ferroptotic cell death in BEAS-2B cells
BEAS-2B cells were exposed to different concentrations of CSE to verify the effect of CSE on bronchial epithelial injury, inflammation, and ferroptosis in COPD. The results showed that CSE obviously triggered cell death (Figures 1(a) and (b)), increased cytotoxicity (Figure 1(c)), and reduced cell viability in a dose-dependent manner (Figure 1(d)). Meanwhile, CSE treatment could increase the level of inflammatory cytokines, such as IL-6 (Figure 1(e)) and TNF-α (Figure 1(f)), in a dose-dependent manner. Then, the characteristic changes in ferroptosis, including lipid peroxidation, GSH depletion, and iron overload, were detected. The results indicated that with the increase in CS concentration, the contents of MDA (Figure 1(g)) and iron (Figure 1(i)) in the BEAS-2B cells increased, while the content of GSH (Figure 1(h)) decreased. Antioxidant stress-related proteins (SLC7A11 and GPX4) and iron metabolism-related proteins (FTH1 and TFR1) are closely associated with ferroptosis. Western blot results indicated that the protein levels of SLC7A11, GPX4, and FTH1 were down-regulated and that of TFR1 was up-regulated by CSE (Figures 1(j) and (k)). These data strongly revealed that CSE could cause cell damage and death, increase the secretion of inflammatory cytokines, and promote ferroptosis in BEAS-2B cells. CSE caused cell damage, inflammation and ferroptosis in BEAS-2B cells. Representative cell morphological changes were observed by phase-contrast microscopy, scale bar = 200 μm (a). Ratio of dead cell count to total cell count in each group related to figure a (b). The cytotoxicity (c) and viability (d) of BEAS-2B cells were measured by LDH and CCK-8 assay. The level of IL-6 (e) and TNF-α (f) in cell supernatant was detected by ELISA. The concentrations of MDA (g), GSH (h), and iron (i) in each group of cells. The protein expression level (j) and semi-quantitative analysis (k) of SLC7A11, GPX4, FTH1, and TFR1 in BEAS-2B cells. The experiment was repeated at least three times. All results are expressed as mean ± SD. *p < 0.05 vs. group of 0% CSE. Error bars in bar charts are standard deviation (SD).
Ferroptosis is involved in cigarette smoke extract-caused cell damage and inflammation in vitro
As expected, the up-regulated levels of intracellular ROS (Figures 2(a) and (b)), MDA (Figure 2(c)), and iron (Figure 2(e)) caused by CSE extensively decreased, and the down-regulated level of GSH (Figure 2(d)) increased after the pretreatment of Fer-1 and DFO. Besides, the protein levels of SLC7A11, GPX4, and FTH1 were significantly up-regulated, and TFR1 was down-regulated in the CSE+Fer-1/DFO group compared with that in the CSE group (Figures 2(f) and (g)). Interestingly, Fer-1 and DFO rescued CSE-caused cell damage and death (Figures 2(h) to (k)), and alleviated the increase in cellular inflammatory cytokines induced by CSE (Figures 2(l) and (m)) in BEAS-2B cells, indicating that Fer-1 and DFO could partially protect epithelial cells from lung injury by suppressing ferroptosis. Fer-1 and DFO alleviated ferroptosis, cell damage and inflammation caused by CSE in BEAS-2B cells. Effects of Fer-1 and DFO on CSE-induced ROS production in BEAS-2B cells (a, b). The intracellular contents of MDA (c), GSH (d), and iron (e) in each group. Western blot results (f) and semi-quantitative analysis (g) of SLC7A11, GPX4, FTH1, and TFR1 in each group of cells. Representative phase-contrast microscopy images (scale bar = 200 μm) (h) and ratio of dead cell count to total cell count (i). The change of cytotoxicity (j) and cell viability (k) in BEAS-2B cells. The level of IL-6 (l) and TNF-α (m) in cell supernatant. The experiment was repeated at least three times. All results are showed as mean ± SD. *p < 0.05 vs. CSE group. Error bars in bar charts are standard deviation (SD).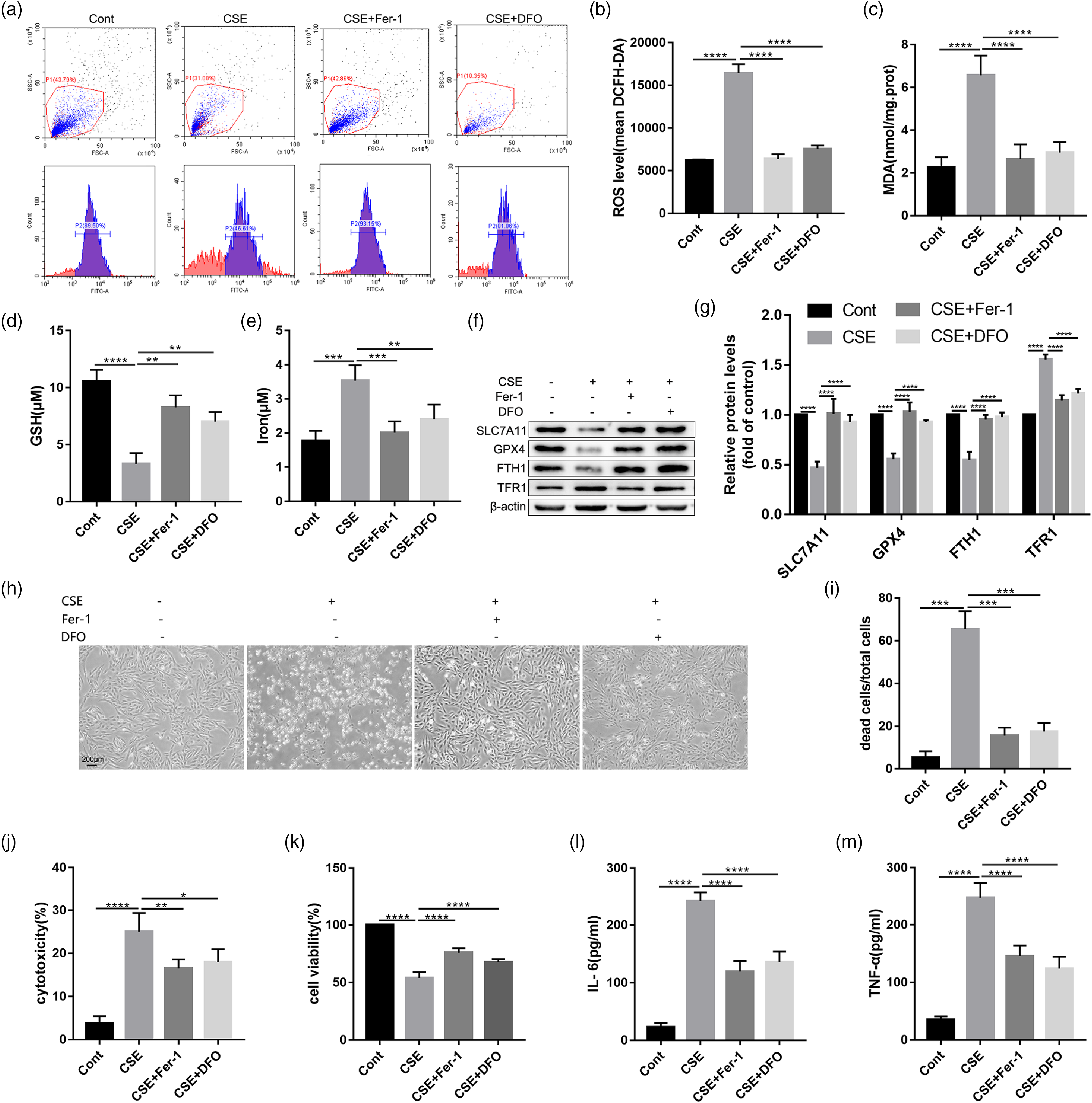
Curcumin inhibits cell damage, inflammation, and ferroptosis-related changes in vitro
BEAS-2B cells were exposed to 2.5% CSE and different concentrations of CUR (5/10/20 μm) to clarify the potential effect of CUR on COPD. The CSE + CUR group had less cell death (Figures 3(a) and (b)), lower cytotoxicity (Figure 3(c)), and higher cell viability (Figure 3(d)) than the CSE group. Moreover, CUR treatment could decrease the levels of IL-6 (Figure 3(e)) and TNF-α (Figure 3(f)). A notable detail that CUR also inhibited characteristic changes in ferroptosis. The levels of MDA (Figure 3(g)), iron (Figure 3(i)), and ROS (Figures 3(j) and (k)) were lower, and the levels of GSH (Figure 3(h)) were higher in BEAS-2B cells treated with CSE and CUR than in those treated with CSE. Furthermore, western blot results displayed that the protein levels of SLC7A11, GPX4, and FTH1 in the CSE + CUR group were up-regulated, and TFR1 was down-regulated compared with those in the CSE group (Figures 3(l) and (m)). These results suggested that CUR could weaken CSE-induced ferroptosis, cellular inflammation, and cell damage in BEAS-2B cells. CUR improved CSE-caused cell damage, inflammation and ferroptosis in BEAS-2B cells. Cell morphological changes (a) and ratio of dead cell count to total cell count (b) in each group. Cytotoxicity (c) and cell viability (d) in BEAS-2B cells. Levels of IL-6 (e) and TNF-α (f) in cell supernatant. The contents of MDA (g), GSH (h), iron (i), and ROS (j and k) in each group. The western blot results (l) and semi-quantitative analysis (m) of SLC7A11, GPX4, FTH1, and TFR1 in BEAS-2B cells. The experiment was repeated at least three times. All results are expressed as mean ± SD. *p < 0.05 vs. CSE group. Error bars in bar charts are standard deviation (SD).
Cigarette smoke-induced inflammation and ferroptosis is attenuated by curcumin in vivo
Data showed that the body weight of rats in the NC group was higher than that in the other three groups, and the body weight in the CS group was lower than that in the other three groups (Figure 4(a)). In addition, compared with the NC group, the alveolar structure was disordered, the alveolar septum was ruptured, and the infiltration of inflammatory cell around the bronchi was increased in the CS group. However, inflammatory cell infiltration was significantly mitigated in the CS + CUR group (Figure 4(b)). CUR also rescued the increased levels of IL-6 (Figure 4(c)) and TNF-α (Figure 4(d)) in the BALF of rats caused by CS. CUR reduced lung epithelial injury, inflammation, and ferroptosis induced by CS in vivo (n = 5). Body weight of rats (a) in NC, CS, CUR, and CS + CUR groups (*p < 0.05 vs. NC group; #p < 0.05 vs. CS group). Morphological changes of lung tissues in each group of rats were examined by H&E staining, scale bar = 100 μm, and arrows represent inflammatory cell infiltration (b). Levels of IL-6 (c) and TNF-α (d) in BALF of rats. The concentrations of MDA (e) and iron (f) in the lung tissues of rats following CS, CUR, and CS+CUR. Western blot showing the expression levels of SLC7A11, GPX4, FTH1, and TFR1 in each group (g and h). All results are showed as mean ± SD. *p < 0.05 vs. CS group. #p < 0.05 vs. CUR group. Error bars in bar charts are standard deviation (SD).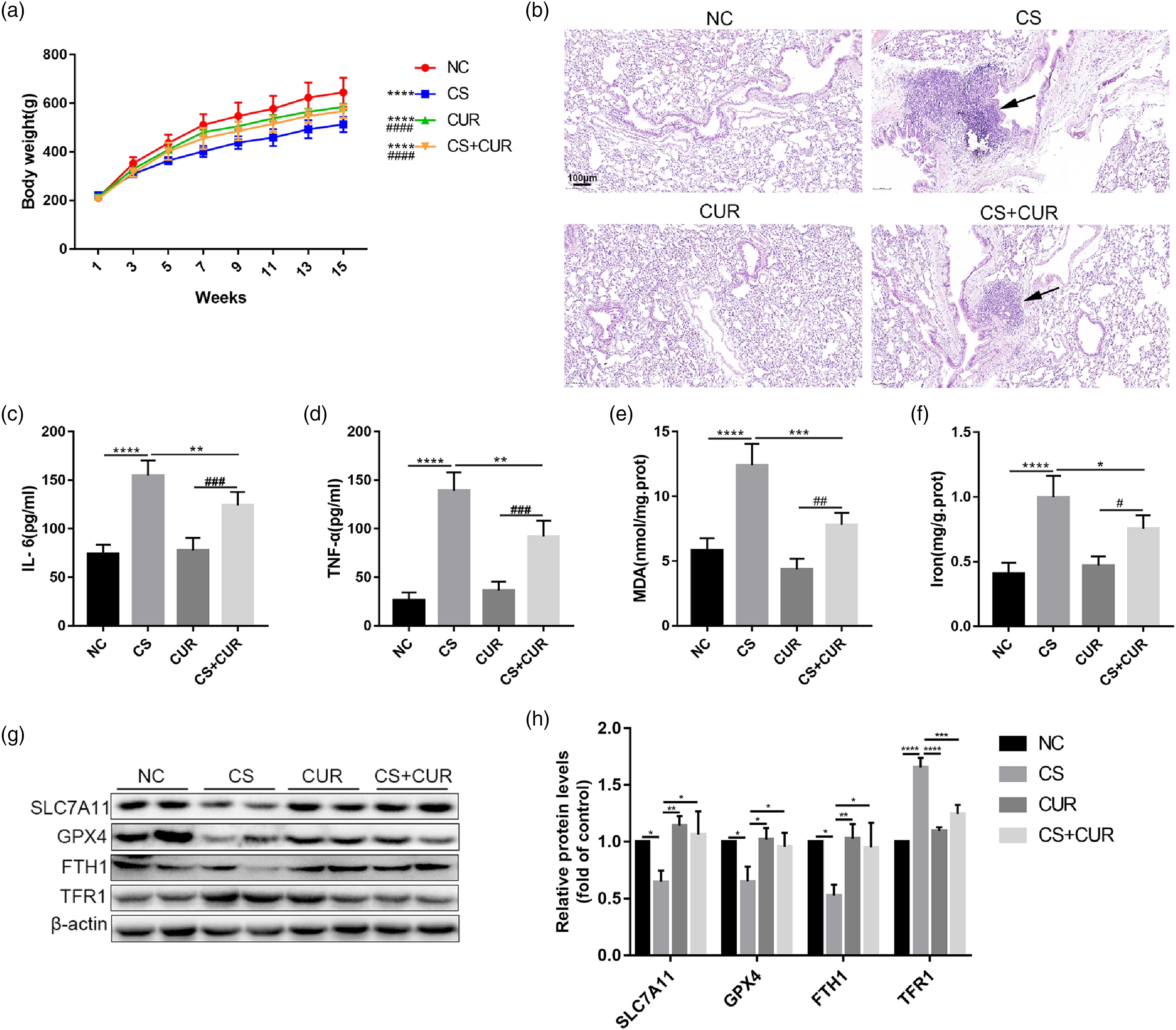
The levels of MDA (Figure 4(e)) and iron (Figure 4(f)) were higher in the lung tissue of the CS group than those in the NC group. Meanwhile, the protein expression levels of SLC7A11, GPX4, and FTH1 decreased, and the level of TFR1 increased in the CS group (Figures 4(g) and (h)). However, after CUR treatment, the ferroptosis-related changes induced by CS were reversed. Collectively, these findings suggested that CUR could inhibit the inflammatory injury of lung, accompanied by the inhibition of ferroptosis.
Discussion
COPD is known as the representative disease caused by CS. 13 Mahalanobish et al. 14 found that CS exposure could activate inflammatory-oxidative stress, trigger ROS accumulation, and reduce GSH/GSSG ratio, which is closely related to the development of COPD. However, decreased antioxidant capacity, increased ROS, lipid peroxidation, GSH depletion, and iron overload is the key events of ferroptosis. 15 Therefore, in the present study, the effect of CSE/CS on ferroptosis in BEAS-2B cells and the lung of rats was explored. The results suggested that CSE could aggravate cell damage and death, increase the level of IL-6 and TNF-α, and induce ferroptotic characteristic changes in BEAS-2B cells. Besides, CS could cause lung injury, increase the infiltration of inflammatory cells and the secretion of inflammatory cytokines, and induce ferroptosis in rats. Some of these findings are consistent with previous studies. For example, Yoshida et al. 3 found that CSE/CS caused ferroptosis-related changes in the lungs of patients with COPD, mouse models, and human bronchial epithelial cells. Furthermore, Park et al. 13 found that CSE may trigger ferroptosis in BEAS-2B cells by endoplasmic reticulum stress and mitochondrial dysfunction.
Fer-1 is a synthetic antioxidant that reduces membrane lipids damage and suppresses lipid peroxidation through a reductive mechanism. 16 DFO, an iron chelator, reduces GSH synthesis and cystine uptake via xCT transporters. 17 Fer-1 and DFO have been proven to be very effective ferroptosis inhibitors.16,17 The present study showed that Fer-1 and DFO not only could inhibit ferroptosis-related changes, but also reduce cell injury, cell death, and cellular inflammation caused by CSE/CS in BEAS-2B cells and lungs of the COPD rat model. This finding suggested that ferroptosis may be closely related to lung injury and inflammatory response in patients with COPD, and the ferroptosis inhibitor plays a potential therapeutic role in COPD by reducing cell death and anti-inflammatory effects. Tsurusaki et al. 18 found that ferroptosis inhibitor decreased the levels of TNF-α, IL-1β, and IL-6; inflammatory cell infiltration; and hepatic cell death in an animal model of steatohepatitis. Kim et al. 19 also found that ferroptotic cell death promotes inflammation by immunogenicity. Ferroptotic cells have stronger immunogenicity due to the release inflammatory cytokines and damage associated with molecular pattern molecules; thus, the cellular environment tends to be in pro-inflammatory state. 19 GPX4 has an important cytoprotective effect through inhibiting lipid peroxidation that leads to inflammation. 20 On the contrary, inflammatory cytokines may induce ferroptosis by reducing the expression of GPX4 in cancer cells. 21 In addition, lipoxygenase could alert immune cells via lysyl oxidase-derived pro-inflammatory metabolites, thereby indirectly inducing ferroptotic cell death. 22
In a previous study, CUR effectively attenuated CSE-associated lung injury, including inflammatory responses and pulmonary function decline.
23
A recent study has demonstrated that CUR could assist in the treatment of renal damage by decreasing ferroptosis.
11
In the present study, CUR attenuated the characteristic changes in ferroptosis, and subsequently improved lung injury and inflammatory response caused by CS in rats. In the CSE-treated BEAS-2B cells, CUR treatment rescued ferroptosis with cell damage and the secretion of inflammatory cytokines. These results inferred a possible protective effect of CUR on COPD by inhibiting ferroptosis (Figure 5). CUR may reduce ferroptosis by inhibiting oxidative stress and lipid peroxidation. Dong et al.
24
found that CUR reduced ferroptosis by activating the nuclear factor E2 related factor 2 (Nrf2)/heme oxygenase-1 (HO-1) signaling pathway. Nrf2 is a vital regulatory factor of cellular redox homeostasis that regulates lipid peroxidation, and it is closely associated with iron storage and transport, which are critical events of ferroptosis. Several target genes were regulated by Nrf2, including intracellular redox-balancing proteins like HO-1, GPX4, and SLC7A11. However, Li et al. found that CUR induced ferroptosis-related changes in breast cancer cells by regulating HO-1.
25
The discrepancy may be associated with the dose of CUR and the type of cell. The proposed model showed that curcumin alleviated CS-induced ferroptosis in the airway epithelium.
Limitations still exist in this work. First, only a single cell line was studied in vitro. Second, in the animal models, lung function, neutrophils, lymphocyte, and other lung injury related indicators were not explored. Finally, the efficacy of CUR in vivo is limited because of its low bioavailability, poor water solubility, and fast metabolism.
In summary, the findings support that ferroptosis plays a crucial role in lung epithelial cell death and inflammatory response caused by CS/CSE in vivo and in vitro, suggesting that the inhibition of ferroptosis has the potential to improve lung injury and alleviate inflammatory response associated with COPD. CUR is an attractive agent in the regulation of ferroptosis, and CUR analog may be prospectively used in the prevention and treatment of COPD.
Footnotes
Acknowledgments
We are very grateful to the Laboratory of Lung Development and Diseases at Nankai University (Tianjin, China) for presenting BEAS-2B cells.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (No. 81670084, 81970084).
Ethical approval
The animal use protocol was approved by the Animal Ethical and Welfare Committee (AEWC) of Tianjin Medical University (Tianjin, China), and the permit number is TMUaMEC2020032.