
Abstract
Sevoflurane is the most commonly used anesthetic in clinical practice and exerts a protective effect on cerebral ischemia-reperfusion (I/R) injury. This study aims to elucidate the molecular mechanism by which sevoflurane postconditioning protects against cerebral I/R injury. Oxygen-glucose deprivation/reperfusion (OGD/R) model in vitro and the middle cerebral artery occlusion (MCAO) model in vivo were established to simulate cerebral I/R injury. Sevoflurane postconditioning reduced neurological deficits, cerebral infarction, and ferroptosis after I/R injury. Interestingly, sevoflurane significantly inhibited specificity protein 1 (SP1) expression in MACO rats and HT22 cells exposed to OGD/R. SP1 overexpression attenuated the neuroprotective effects of sevoflurane on OGD/R-treated HT22 cells, evidenced by reduced cell viability, increased apoptosis, and cleaved caspase-3 expression. Furthermore, chromatin immunoprecipitation and luciferase experiments verified that SP1 bound directly to the ACSL4 promoter region to increase its expression. In addition, sevoflurane inhibited ferroptosis via SP1/ACSL4 axis. Generally, our study describes an anti-ferroptosis effect of sevoflurane against cerebral I/R injury via downregulating the SP1/ASCL4 axis. These findings suggest a novel sight for cerebral protection against cerebral I/R injury and indicate a potential therapeutic approach for a variety of cerebral diseases.
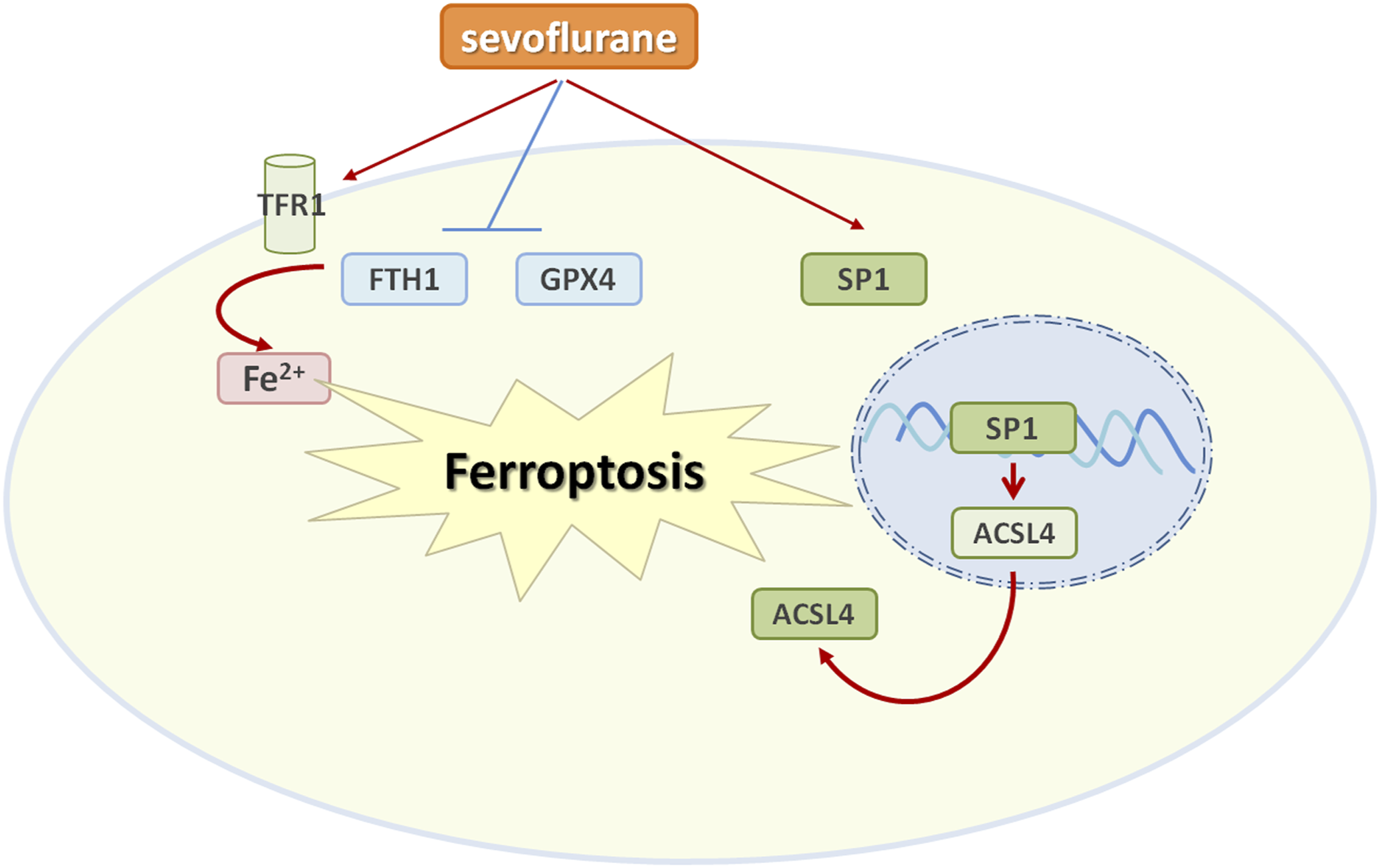
Introduction
Ischemic stroke is the most common neurological disease, accounting for 85% of all stroke cases, and is the main cause of devastating cerebrovascular disease. Timely ischemia-reperfusion can alleviate or avoid prevent neurological defects and damage. However, it may also exacerbate neuronal death and neurological dysfunction. 1 Cerebral I/R injury is a complex pathophysiological process, including inflammation, oxidative stress, edema, and neuronal death.2–4 Ferroptosis is an iron-dependent novel programmed cell death, characterized by the accumulation of lipid hydrogen peroxide to lethal levels. 5 Recent findings reveal that ferroptosis is a major driver of I/R injury and ferroptosis inhibitors have successfully prevented or reduced I/R injury in various organs. 6 Inhibition of ferroptosis may be an effective treatment strategy for cerebral ischemic injury and may help to reduce cell death during cerebral I/R injury. 7
Many researchers have explored the efficacy and the underlying mechanisms of many drugs and treatment strategies for cerebral I/R injury.8,9 Sevoflurane is the most commonly used anesthetic in clinical practice. In addition, it has been shown to have anti-inflammatory and neuroprotective activities. It has been reported that sevoflurane reduced neurological impairment, cerebral infarction volume, and inflammatory cytokine levels.10–12 At the same time, sevoflurane has been shown to reduce neuronal apoptosis and antioxidant stress.13–15 Sevoflurane plays a protective role in neuronal HIBI by inhibiting apoptosis and necrosis. 16 Sevoflurane has been reported to be an effective neuroprotective agent in cerebral I/R injury. Sevoflurane conditioning can protect against brain injury induced by middle cerebral artery occlusion in rats by inhibiting autophagy and apoptosis. 14 Evidence suggests that general anesthesia induced by sevoflurane may be associated with cognitive development and neurodegeneration. 17 Studies have shown that GA induced by sevoflurane inhalation disrupts iron homeostasis and leads to iron overload in hippocampal neuron culture in vitro and hippocampus in vivo. 18 Sevoflurane exposure during the third trimester resulted in significantly reduced Nrf2 expression and increased ROS accumulation in embryonic neural stem cells, reduced neurogenesis, and promoted ferroptosis in the embryonic prefrontal cortex. 19 However, the relationship between sevoflurane and ferroptosis in cerebral I/R has not been covered.
Specificity protein 1 (SP1) belongs to the specific protein/Kruppel-like factor family, which also includes SP2, SP3, and SP4. Emerging evidence suggests that SP1 is an important regulator of a variety of cellular events, including cell cycle, proliferation, and apoptosis.20,21 SP1 is involved in the development of neurological diseases. 22 Sevoflurane-induced SP1 upregulation affects the activation of cholinergic anti-inflammatory pathways, leading to increased neuroinflammation and apoptosis. 23 Growing studies showed that SP1 shaped ferroptosis sensitivity via either transcription-dependent or -independent manners. 24 For instance, SP1 binds to SP1 response elements in the Prdx6 promoter, directly upregulates the expression of Prdx6 in podocytes, and then protects podocytes against diabetic nephropathy by mitigating ferroptosis. 25 Inhibition of SP1 repressed ferroptosis of endothelial cells and retarding the occurrence of atherosclerosis. 26 However, the role of SP1 in sevoflurane-mediated neuroprotection against cerebral I/R injury remains unclear.
In this study, we found that sevoflurane alleviated cerebral I/R injury and suppressed ferroptosis in MCAO rats. Furthermore, sevoflurane remarkably decreased the expression of SP1 in MCAO rat brain and OGD/R-treated HT22 cells. And, SP1 overexpression could attenuate the neuroprotective effect of sevoflurane in OGD/R-treated HT22 cells. Mechanistically, SP1 increased ACSL4 expression at the transcriptional level and then activated ferroptosis events. Our study will provide a new perspective for preventing I/R injury.
Materials and Methods
Cell Culture
Mouse hippocampal neuron HT22 cells were obtained from Procell (Wuhan, China). HT22 cells were cultured with HT22 cells Special medium (Procell, Wuhan, China) in a humidified atmosphere of 37°C and 5% CO2.
Oxygen-Glucose Deprivation (OGD/R) Model
HT22 cells were cultured with glucose-free DMEM in a triple-gas incubator (N2:O2:CO2 = 94:1:5) for 6 h. Then, HT22 cells were incubated in a high-glucose and serum-free DMEM medium at 95% air and 5% CO2 at 37°C for 24 h.
Cell Transfection
The SP1-expressing vectors and small interfering RNAs (siRNAs) targeting SP1 were provided by GenePharma (Shanghai, China). All siRNAs and vectors were transfected in HT22 cells using Lipofectamine 3000 (Invitrogen, USA) for 48 h before OGD/R treatment.
Cell apoptosis analysis
Cell apoptosis was analyzed by Annexin V-FITC and PI staining and flow cytometry analysis using the TransGen Biotech V-FITC/PI Apoptosis Assay Kit (Beijing, China). Cells were collected, stained with Annexin V-FITC and PI, and immersed in darkness for 15 min. Cell apoptosis was analyzed by flow cytometry.
Quantitative Real-Time polymerase chain reaction (QRT-PCR)
The primer sequences used in this study.

Animals and Groups
Adult male SD rats (250–300 g) were purchased from Charies River (Beijing, China). The animals were placed in laboratory cages, kept on a 12-h light-dark cycle, and had free access to food and water throughout the study. The rats were randomly assigned to the sham (only the left neck was exposed without ligation) group, MACO group, and sevo + MACO (2.5% sevoflurane before refusion) group. The MCAO model was made by a modified nylon suture method. After 1 h of ischemia, the suture was gently pulled to the beginning of the external carotid artery and re-perfused for 24 h. For sevoflurane postconditioning, rats were stabilized in a gas-tight anesthesia chamber with sevoflurane inhalation for 1 h at the onset of blood refusion. Sevoflurane (AbbVie, Japan) was delivered at a concentration of 2.5% through a vaporizer (Vapor 2000, Germany). In the sham or MCAO group, rats were only exposed to the mixed gas (95% O2 and 5% CO2).
Evaluation of neurobehavioral deficits
After reperfusion for 24 h, neurobehavioral behavior deficits were assessed with the modified neurological severity score (mNSS) test as previously described. 27 mNSS ranged from 0 to 18 (Normal score was 0; Maximum defect score: 18). A score of 13–18 indicates severe injury, 7 to 12 indicates moderate injury, and 1 to 6 indicates mild injury.
Evaluation of infarct volume
The rats were sacrificed with an injection of sodium pentobarbital, and the brains were immediately removed. Then, rat brains were cut into six coronal sections with 2 mm thick. Sections were stained with 2% TTC solution f at 37°C or 15 min. The total brain sections and the infarcted areas were quantified by Image J software. Infarct volume (%) = infarct volume on the infarct side/normal hemisphere volume *100%.
TUNEL staining
The rat brains were dehydrated and embedded in paraffin, then cut into 4 μm coronal sections. The sections were then dewaxed and hydrated for terminal deoxynucleotide transferase (TdT) -mediated dUTP notched-end labeling (TUNEL) staining. Neuronal apoptosis was detected using One-step TUNEL In Situ Apoptosis Kit (Elabscience, Wuhan, China) following to manufacturer’s instructions.
Western blotting
The peri-infarcted cerebral cortex is lysed in a lysis buffer. Bradford assay was performed to determine the protein concentration. The sample containing 40 μg protein was fractal by 10% SDS-polyacrylamide gel electrophoresis and electroplated onto PVDF membrane. The bands were scanned. Beta-actin was used as an internal control. The antibodies information: anti-SP1 (1:1000; Protein technology), anti-ASCL4 (1:1000; Protein technology), anti-cleaved caspase 3 (1:1000; Protein technology), anti-GPX4 (1:1000; Protein technology), anti-FTH1 (1:1000; Protein technology), anti-TFR1 (1:1000; Protein technology), anti-β-actin (1:2000; Protein technology).
Statistical Analysis
All data analysis was performed using GraphPad Prism 8 (GraphPad Software, San Diego, USA). The data were expressed as SD ± mean, and the data of different groups were compared by one-way ANOVA. p < 0.05 was considered statistically significant.
Results
Sevoflurane postconditioning reduces neurological deficits and cerebral infarction after I/R
To investigate the potential effect of sevoflurane on I/R injury, the MCAO rats were treated with sevoflurane before refusion. Behavioral tests were performed at 24 h after MCAO. neurological deficit evaluation showed that the mNSS score in the MCAO group was significantly increased compared to the sham group. However, sevoflurane postconditioning significantly reduced the neurological deficit score (Figure 1(A)). Moreover, TTC pathological examination displayed that MACO rats showed significant cerebral infarction, and sevoflurane postconditioning remarkably reduced the infarct size compared with the MACO group (Figure 1(B)). Furthermore, HE staining showed normal neurons with regular morphology in the sham group. However, reduced neurons and short neurite were observed in MACO rats. However, sevoflurane postconditioning significantly attenuated this histopathological damage (Figure 1(C)). TUNEL staining confirmed that MACO rats showed obvious cell apoptosis, while sevoflurane postconditioning decreased the apoptosis (Figure 1(D)). This was also confirmed by Western blotting analysis of cleaved caspase-3 (Figure 1(E)). These results show that sevoflurane can reduce I/R-induced cerebral damage. Sevoflurane postconditioning reduces neurological deficits and cerebral infarction after I/R. (A) Nervous system behavior was assessed with the mNSS test (n = 12). (B) The infarct volume was evaluated using TTC staining (n = 6). (C) Photomicrograph of the rat brain showing the histologic structure (H&E scale bar 20 μm). (D) TUNEL was performed to assess cell apoptosis (n = 3). (E) Western blot was used to determine the expression of cleaved caspase 3 (n = 3). ***p < 0.001.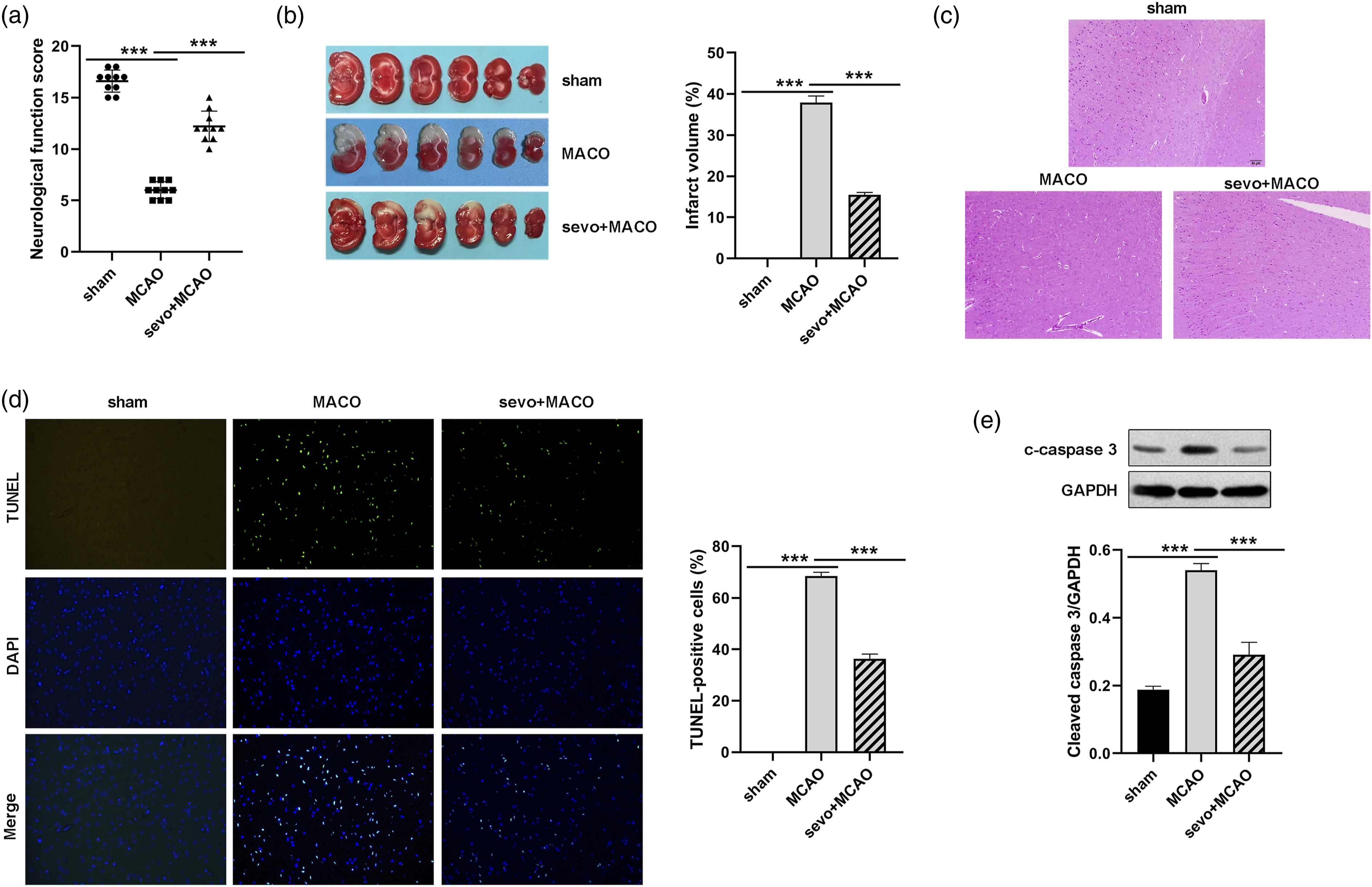
Sevoflurane postconditioning reduces ferroptosis induced by cerebral I/R
Accumulating pieces of evidence reveal that ferroptosis is a major driver of I/R injury. In this study, we found that iron levels were increased in the brain tissues of MCAO rats, and sevoflurane postconditioning reduced the elevated iron levels compared with the MCAO group (Figure 2(A)). Furthermore, we measured the expression levels of ferroptosis-related markers including ACSL4, GPX4, transferrin receptor (TFR1), and FHT1 Western blot analysis showed that the negative regulators of ferroptosis ACSL4 and GPX4 were decreased and the positive regulators of ferroptosis TFR1 and FHT1 were increased in the brain tissues of MCAO rats, and sevoflurane postconditioning reversed the expression of these markers (Figure 2(B)). These findings indicate that sevoflurane can inhibit I/R-induced ferroptosis. Sevoflurane postconditioning reduces ferroptosis induced by cerebral I/R. (A) The iron amount was measured in the brain tissues of rats (n = 6). (B) Western blot was performed to determine the expression levels of ACSL4, FHT1, TFR1, and GPX4 in the brain tissues of rats (n = 3). ***p < 0.001.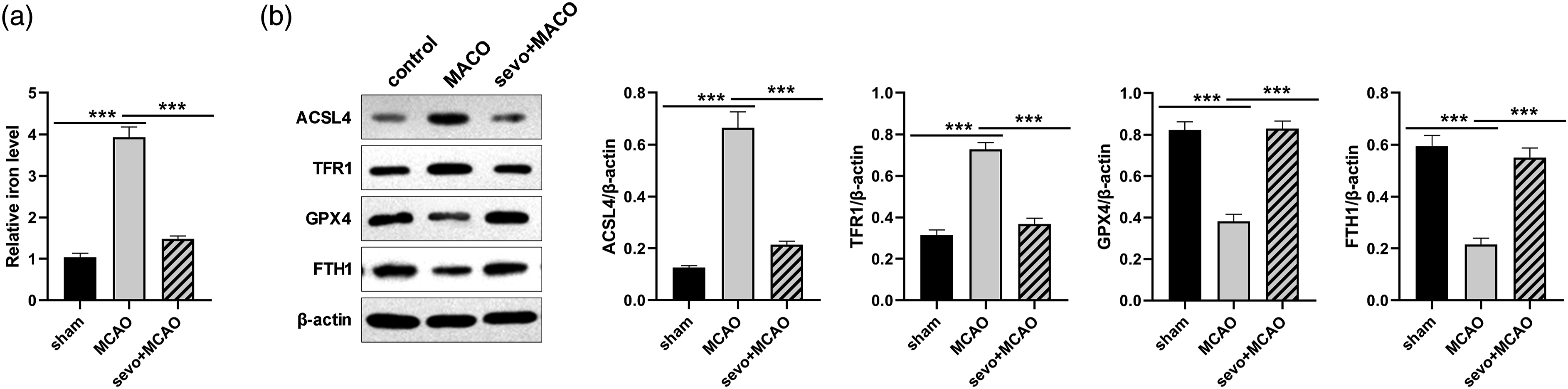
Sevoflurane postconditioning exerts neuroprotection via downregulating SP1 expression
Previous studies have shown that SP1 was an important regulator in neurological diseases, and is also associated with susceptibility to ferroptosis. Therefore, we speculated that sevoflurane may exert neuroprotection by regulating SP1-mediated ferroptosis in the MCAO model. QRT-PCR and Western blot results showed that the expression levels of SP1 were increased in the MCAO group compared with the sham group but decreased significantly after sevoflurane treatment (Figures 3(A) and (B)). Moreover, HT22 cells were treated with OGD/R and sevoflurane. QRT-PCR and Western blot results showed that SP1 was highly expressed in OGD/R-induced HT22 cells and sevoflurane treatment significantly inhibited its expression (Figures 3(C) and (D)). Sevoflurane postconditioning exerts neuroprotection via downregulating SP1 expression. (A) RT-qPCR and (B) Western blot were used to determine the expression levels of SP1 in the brain tissues of rats with MCAO or/and sevo. (C) RT-qPCR and (D) Western blot were used to determine the expression levels of SP1 in HT22 cells treated with OGD/R or/and sevo. (E) RT-qPCR and (F) Western blot was used to determine the expression levels of SP1 in HT22 cells transfected with SP1-expressing plasmids (SP1_OE) or Vector. (G) Cell viability was detected in HT22 cells using a CCK-8 assay. (H) Cell apoptosis was analyzed in HT22 cells using Annexin V-FITC and PI staining and flow cytometry analysis. (I) Western blots of cleaved caspase three in each group. n = 3. ns, no significance; **p < 0.01, ***p < 0.001. sevo, sevoflurane.
To investigate the role of SP1 during the treatment of cerebral I/R injury with sevoflurane, HT22 cells were transfected with pcDNA3.1-SP1 plasmids to induce SP1 overexpression in HT22 cells, and the transfected HT22 cells were then treated with OGD/R and sevoflurane. Transfection with pcDNA3.1-SP1 plasmids significantly enhanced SP1 expression in HT22 cells (Figures 3(E) and (F)). Furthermore, the viability and apoptosis of modified HT22 cells were estimated by CCK-8 and flow cytometry. Overexpression of SP1 partially eliminated the protective effect of sevoflurane on OGD/R cells, which showed decreased cell viability (Figure 3(G)) and increased cell apoptosis (Figure 3(H)). Besides, the expression of cleaved caspase-3 in HT22 cells was examined by Western blotting. As shown in Figure 3(i), sevoflurane treatment reduced the expression of cleaved caspase-3 in OGD/R-treated cells. And, the expression of cleaved caspase-3 was significantly increased in SP1-overexpressing HT22 cells. These data suggest that sevoflurane postconditioning exerts neuroprotection via downregulating SP1 expression.
Transcription factor SP1 binds to the ACSL4 promoter and promotes its expression
Acyl-CoA synthetase long-chain family member 4 (ACSL4) has been reported to serve as a major player in the progress of ferroptosis in various diseases. And previous studies have identified ACSL4 as a target gene for SP1. Therefore, we investigated whether SP1 regulates ACSL4 transcription. As shown in Figures 4(A) and (B), the knockdown of SP1 decreased SP1 and ACSL4 mRNA and protein levels, while SP1 overexpression increased SP1 and ACSL4 mRNA and protein levels. These results suggest that SP1 positively regulates ACSL4 expression in HT22 cells. To evaluate whether SP1 affects the promoter activity of ACSL4, luciferase, and ChIP experiments were conducted. A segment in the ACSL4 promoter was identified as two putative targets (relative to TSS −221 to −229 bp and −83∼-91 bp) via the JASPAR database and Figure 4(C) showed the putative SP1 binding sites in the ACSL4 promoter. ChIP assay showed that the P1 region (+100∼-200) and P2 region (−200∼-500) of ACSL4 promoter were enriched in the SP1 immunoprecipitation complex, suggesting that SP1 binds to these two regions of the ASCL4 promoter. Then, we constructed luciferase plasmids containing ACSL4 wild-type (ACSL4-WT) or deleted corresponding sites (ACSL4-MUT1, ACSL4-MUT2, and ACSL4-MUT2). These luciferase plasmids were co-transfected with pcDNA3.1-SP1 or negative vector to perform the dual-luciferase reporter gene assay. As shown in Figure 4(D), SP1 overexpression significantly increased the luciferase activity of the WT group. Interestingly, SP1 overexpression promoted the luciferase activity of the two single region-deletion plasmids, but did not affect the luciferase activity of both sequence-deletion plasmids, indicating SP1 can bind to the two regions of the ASCL4 promoter. Transcription factor SP1 binds to ACSL4 promoter. (A) RT-qPCR and (B) Western blot were used to examine the expression levels of SP1 and ACSL4 in HT22 cells after SP1 was overexpressed or silenced. (C) The JASPAR website predicts the binding of transcription factor SP1 to the ACSL4 promoter. (D) ChIP assay was employed to assess the binding ability of SP1 and ACSL4 promoter. (E) The luciferase reporter assay was conducted to verify the binding activity of SP1 to the ACSL4 promoter. n = 3. **p < 0.01, ***p < 0.001.
Sevoflurane postconditioning inhibits ferroptosis by inhibiting SP1/ACSL4 axis
HT22 cells were transfected with pcDNA3.1-SP1 plasmids and then treated with OGD/R and sevoflurane. Western blot analysis showed that Sevoflurane postconditioning reduced decreased ACSL4 and TFR1 and increased GPX4 and FHT1 in OGD/R-treated HT22 cells. However, these effects were reversed by SP1 overexpression (Figure 5(A)). The intracellular iron levels showed the same trend as TFR1 in each group (Figure 5(B)). Sevoflurane postconditioning inhibits ferroptosis via inhibiting SP1/ACSL4 axis. (A) Western blot was used to determine the expression levels of ACSL4, TFR1, GPX4 and FHT1. (B) The levels of iron were determined using commercial kits. **p < 0.01. ***p < 0.001.
Discussion
Sevoflurane is an effective neuroprotective agent for cerebral I/R injury.12,13 Sevoflurane treatment has been reported to reduce neurological impairment, cerebral infarction volume, and inflammatory cytokine levels. Sevoflurane has also been shown to reduce neuronal apoptosis and antioxidant stress.28,29 Sevoflurane protects against brain injury in several ways. Therefore, a comprehensive understanding of its neuroprotective effect will help open up new therapeutic approaches for I/R and provide the theoretical basis for new clinical treatment decisions. In this study, we demonstrated that sevoflurane post-treatment plays a neuroprotective role during I/R by down-regulating SP1 expression. Investigating the mechanism, we found that SP1 as a transcription factor promotes the transcriptional activity of ASCL4, a positive regulator of ferroptosis. We confirmed that sevoflurane treatment reduced intracellular iron accumulation by inhibiting the SP1/ASCL4 axis, thereby alleviating cerebral I/R injury. In conclusion, our study elucidates the neuroprotective effect of sevoflurane from a new perspective of iron homeostasis and provides the basis for the potential clinical use of its clinical application in cerebrovascular diseases.
Ferroptosis is an iron-dependent non-apoptotic programmed cell death process, characterized by an increased lipid peroxidation that leads to cell death by disrupting the integrity of cell membranes. 30 Evidence suggests that ferroptosis plays a key role in cerebral I/R injury and inhibition of ferroptosis plays a neuroprotective role. 31 In addition, iron deposition in brain tissue have also been observed in animal models of cerebral I/R and many neuroprotective drugs alleviate cerebral I/R injury by inhibiting ferroptosis.32–34 For example, γ-glutamylcysteine decreases cerebral I/R injury through suppressing ferroptosis. 35 Calycosin exerts neuroprotection effects against cerebral I/R injury by inhibiting ACSL4-dependent ferroptosis. 36 Consistent with these reports, in this study, sevoflurane was found to inhibit the occurrence of ferroptosis events in MCAO rat brain and OGD/R-treated HT22 cells, which were represented by reduced iron content, decreased expression of TFR1 and FHT1, and increased expression of ACSL4 and GPX4. However, previous studies have shown that sevoflurane has different effects on ferroptosis in different cells and conditions. For instance, sevoflurane induces ferroptosis in glioma cells and embryonic prefrontal cortex.19,37 On the contrary, sevoflurane inhibits ferroptosis in lipopolysaccharide-induced acute lung injury. 38 We speculate that this phenomenon may be related to cell background and sevoflurane administration procedures including dose, administration mode and duration. Collectively, sevoflurane decreases cerebral I/R injury through suppressing ferroptosis.
Accumulated evidence suggests that SP1 is a novel molecular target that regulates the pathogenesis of I/R injury. SP1 has previously been assessed for association with central nervous system disease. For example, Wang et al. 39 reported that SP1 was expressed in brain tissue, and the increase of SP1 decreased the survival rate of neurons. In addition, SP1 is upregulated in neuropathological developments, including PD and Alzheimer’s disease.40,41 Furthermore, Qin et al. 42 showed that inhibition of SP1 activation effectively inhibits OGD/R-induced microglial inflammatory activation. Recent evidence suggests that SP1 expression is increased in ischemic/hypoxic conditions. Together, these data reveal the key influence of SP1 in neuropathology. In this work, we found that SP1 was down-regulated in OGD/R-treated HT22 cells and MCAO rat brain tissue. In addition, SP1 overexpression weakened the protective effect of sevoflurane on OGD/R-treated cells, showing reduced cell viability and increased apoptosis and apoptosis marker protein Cleaved caspase three expressions, compared with the sevoflurane alone treatment group. Meanwhile, overexpression of SP1 attenuated the neuroprotective effect of sevoflurane on MCAO rats, including decreased neurological function score, increased infarct size, increased apoptosis, and apoptosis marker protein Cleaved caspase three expressions, compared with sevoflurane alone. Based on these data, we conclude that sevoflurane plays a neuroprotective role in cerebral I/R injury by down-regulating SP1.
SP1 is a classic and well-studied transcription factor that regulates the transcription of many genes by binding to GC-box elements in the promoter region, thereby contributing to disease progression.43,44 It was found that SP1 protects neurons, glial cells, and endothelial cells by promoting the transcription of several antioxidant proteins, including zinc finger protein 179, and various antioxidant enzymes, and affecting ion transporters on the cytoplasmic side.24,45,46 Previous studies have reported that SP1 upregulates ACSL4 expression by binding to the ACSL4 promoter region. 47 In this study, we identified ACSL4 as a direct target gene for SP1. Overexpression or knockdown of SP1 promoted or inhibited its expression. Luciferase and ChIP assay confirmed that Sp1 promotes ACSL4 transcriptional activity. In addition, ACSL4 expression was increased in the OGD/R cell model and MCAO rat model, but decreased after sevo treatment, while overexpression of SP1 promoted its re-expression. ACSL4 is a key enzyme that regulates lipid composition and has been identified as a key molecule of ferroptosis. 48 ACSL4 can enrich ω 6 fatty acids, such as arachidonic acid and adrenic acid to cellular membranes. Compared with ω three fatty acids, ω 6 fatty acids increased the susceptibility of cells to ferroptosis, It has been shown that genetic or pharmacological inhibition of ACSL4 attenuates the occurrence of ferroptosis ACSL4-mediated ferroptosis has been reported to promote tissue injuries, such as intestinal I/R injury and brain ischemic injury.49–51 In this study, by analyzing the key proteins in the regulation of ferroptosis, FTH1, ACSL4, and GPX4, as well as the intracellular iron ion levels, we found that SP1 overexpression promoted ACSL4 expression and weakened the inhibition of ferroptosis by sevo. In conclusion, our study suggests that sevo treatment inhibits ferroptosis events by inhibiting the SP1/ASCL4 axis, thereby reducing cerebral I/R damage.
Conclusions
In summary, our findings demonstrated that sevoflurane effectively suppressed ferroptosis and alleviated cerebral I/R injury via inhibition of the SP1/ASCL4 signal axis. This study provides favorable evidence for the application of sevoflurane in the treatment of cerebral I/R damage.
Footnotes
Author Contributions
NL and XL contributed to the study conception and design, material preparation, data collection, and analysis. NL wrote the first draft of the manuscript. XL read and approved the final manuscript.
Declaration of conflicting interests
The authors have no relevant financial or non-financial interests to disclose.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.”
Ethical approval
The animal study was reviewed and approved by the Animal Ethics and Welfare Committee of the Institute of Radiation Medicine, Chinese Academy of Medical Sciences (No. IRM-DWLL-2021190).
Data availability
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.