
Abstract
Purpose:
Cerebral ischemia is the main cause of permanent adult disabilities worldwide. This study investigated the reparative effects and potential mechanisms of methylphenidate (MPH), a medication for the treatment of attention-deficit/hyperactivity disorder.
Methods:
In vitro oxygen-glucose deprivation/reperfusion (OGD/R) and in vivo cerebral ischemia-reperfusion models were established. Sprague-Dawley (SD) rats were randomly divided into four groups (n = 20): Sham, Model, and MPH (0.5 and 1 mg/kg). Rats in MPH groups were treated with 0.5 or 1 mg/kg MPH via intraperitoneal injection for 7 days. Rats in the Sham and Model groups were treated with PBS during the same period. Cell viability was measured using MTT assay. Apoptosis was detected by Annexin V/PI staining. Protein expression was detected by Western blot. The volume of cerebral infarction was detected by triphenyltetrazolium chloride (TTC) staining. The DNA damage in ischemic brain tissues was detected by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay.
Results:
MPH treatment significantly reduced OGD/R-induced cell damage, shown by the increased cell viability and decreased apoptotic rate. p-AMPK and p-ACC protein expression increased in the OGD/R model after MPH treatment. The addition of AMPK inhibitor largely abolished the neuroprotective effects of MPH, evidenced by the reduced cell viability, increased apoptotic rate, and decreased protein expression of p-AMPK as well as p-ACC. Moreover, MPH treatment significantly alleviated the cerebral ischemia-reperfusion injury and decreased apoptosis in brain tissues, which may be associated with the AMPK/ACC pathway.
Conclusions:
MPH exerted protective activities against oxidative stress in the OGD/R model and ameliorated brain damage of rats in the middle cerebral artery occlusion model, at least in part, through activating the AMPK pathway. These data demonstrated neuroprotective properties of MPH and highlighted it as a potential therapeutic agent against cerebral ischemia-reperfusion injury.
Keywords
Introduction
As a common vascular disease in the central nervous system (CNS), ischemic stroke is one of the most common causes of death and long-term disability worldwide.1,2 Of the 15 million people who have a stroke each year, more than 5 million die and a further 5 million are left permanently disabled. Ischemic stroke caused by sudden blood vessel occlusion with an embolism or thrombus accounts for approximately 80% of all stroke cases. 3 There is overwhelming evidence that apoptosis contributes significantly to cell death subsequent to cerebral ischemia-reperfusion (I/R), in addition to necrosis.4,5 Pathogenic mechanisms following cerebral I/R are associated with elevation of the intracellular Ca2+ level, energy failure, release of excitatory neurotransmitters, inflammation, and oxidative stress. 6 However, the exact mechanism has not been established.
One of the fundamental requirements for all cells is to balance ATP generation and consumption. AMP-activated protein kinase (AMPK), which is a highly conserved sensor of intracellular adenosine nucleotide levels and activated when intracellular ATP levels are low, plays a critical role in regulating growth and reprogramming metabolism. 7 AMPK has a protective role in myocardial ischemia, angiogenesis, diabetic cardiac injury, and fluid shear stress.8–11 Acetyl coenzyme A carboxylase (ACC) is an enzyme catalyzing the conversion of acetyl coenzyme A to malonyl coenzyme A, which is involved in the energy metabolism of fatty acids in animals as a promising target for metabolic syndrome intervention. ACC has been demonstrated as a potential immune modulatory target of cerebral ischemic stroke.12,13 Previous studies showed that energy metabolism dysfunction plays a significant role in ischemic stroke. AMPK/ACC resists cerebral ischemic injury by switching on catabolic pathways and switching off ATP-consuming processes, which may be a potent target for ischemic stroke treatment.14,15
Cultured cells subjected to hypoxia, fuel deprivation and re-oxygenation replicate the features of I/R. Thereafter, an oxygen and glucose deprivation-reperfusion (OGD/R) cell model is commonly used to simulate the state of neuronal I/R injury that results in neuronal apoptosis. 16 Many animal stroke models have focused on the middle cerebral artery (MCA) because most ischemic strokes occur in the MCA territory. The intraluminal monofilament model of middle cerebral artery occlusion (MCAO) involves the insertion of a surgical filament into the external carotid artery and threading it forward into the internal carotid artery (ICA) until the tip occludes the origin of the MCA, resulting in a cessation of blood flow and subsequent brain infarction in the MCA territory. This technique can be used to model transient or permanent occlusion. 17
Currently, there are two therapeutic strategies applied to cerebral I/R injury. The best treatment is to restore blood supply by administering thrombolytic agents. However, thrombolytic therapy is limited by a very narrow therapeutic window and can also lead to hemorrhagic complications. 18 Neuroprotection, as the other strategy, has attracted more attention in recent years. Methylphenidate (MPH) is a dopamine-reuptake inhibitor approved for the treatment of attention-deficit/hyperactivity disorder (ADHD). Effects of MPH on the nervous system are controversial due to the dosage. High doses (10 or 20 mg/kg) caused mitochondrial dysfunction, oxidative stress and inflammatory changes in brain cells and induced neurodegeneration in the hippocampus and cerebral cortex of rats.19–21 However, at a low concentration, MPH exerted neuroprotective activities against oxidative stress in vitro.22,23 This study was to investigate the neuroprotective effects of MPH in OGD/R and MCAO rat models, and further explore the underlying mechanisms.
Materials and methods
Chemicals and reagents
3-(4,5-dimethylthiazol)-2,5-diphenyltetrazolium-bromide (MTT) was purchased from Sigma–Aldrich (St. Louis, MO, USA). Caspase-3 and caspase-9 activity assay kits were purchased from R&D Systems (Minneapolis, MN, USA). The TUNEL assay kit was purchased from Roche (Mannheim, Germany). The Annexin V/PI apoptosis assay kit was purchased from ThermoFisher (Waltham, MA, USA). All solvents and chemicals were of analytical grade and purchased from Sinopharm Co. Ltd. (Shanghai, China). The AMPK inhibitor Dorsomorphin (purity: 99.65%) was purchased from MedChemExpress (Monmouth Junction, NJ, USA). Methylphenidate hydrochloride was purchased from the National Institutes for Food and Drug Control (Beijing, China) and dissolved in phosphate-buffer saline (PBS).
Cell culture and oxygen-glucose deprivation/reperfusion (OGD/R)
Primary neurons were isolated from newborn Sprague-Dawley (SD) rats (Vital River Ltd., Beijing, China). The hippocampal tissues were firstly digested with 0.125% trypsin for 10 min. After separation and centrifugation, neurons were inoculated into 6-well plates treated by poly-d-lysine (Sigma–Aldrich, St. Louis, MO, USA) overnight. Cells were maintained with neurobasal medium containing 2% B27, 1% glutamine and cytarabine at 37°C. 24 To establish the oxygen-glucose deprivation (OGD) model, the culture medium was removed and cells were washed twice using RPMI1640 medium without glucose. Cells were then transferred to 37°C, N2/CO2 (95%/5%) in a hypoxic incubator chamber (Thermo Fisher, Waltham, MA, USA) for 10 min. RPMI1640 medium without glucose was added to cells for 2 h in the hypoxic incubator chamber (OGD phase). Next, cells were cultured in RPMI1640 medium containing 7.5% horse serum and 10% FBS in an air/CO2 (95%/5%) incubator for 24 h (reperfusion [R] phase). Control cells were cultured under normoxic conditions without OGD treatment. MPH at different concentrations (1, 3, and 10 µM) was added 2 h prior to OGD. The concentrations of MPH were based on our preliminary study and literatures.22,23 To further validate the MPH mechanism of action, the specific inhibitor of AMPK (10 µM Dorsomorphin) was added 2 h before MPH treatment.
Cell viability assay
The cell viability was assessed using MTT assay. After treatment, cells were incubated with 0.5 mg/mL MTT at 37°C for 1 h. The formazan was dissolved in DMSO and the absorbance was determined at 570 nm with a microplate reader. 25
Apoptosis analysis
After treatment, 2 × 10 6 cells were collected and re-suspended in 500 µl of binding buffer with 5 µl of Annexin V-FITC/PI for 10 min in the dark. Apoptosis was detected with the FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA). Data were analyzed using FlowJo V10.
Rat ischemic/reperfusion model and treatment
One hundred and twenty male SD rats, weighing 260–280 g, were purchased from Vital River Ltd. (Beijing, China). Rats were maintained in a temperature-controlled (23 ± 2°C) and humidity-controlled (40–70%) facility under specific pathogen-free condition. Rats had access to food and water ad libitum. The Ethics Committee of Hunan Provincial Brain Hospital has reviewed and approved this study. All experimental procedures were carried out according to the Chinese legislation on the Use and Care of Animals.
The middle cerebral artery occlusion (MCAO) method was used to establish the cerebral I/R injury.26,27 Rats were anesthetized with intraperitoneal injection of a mixture of 50 mg/kg ketamine and 10 mg/kg xylazine. The common left carotid artery was exposed and isolated, while the middle cerebral artery was occluded by inserting a nylon filament (0.24–0.28 mm diameter) into the internal carotid artery. This was advanced 18–20 mm past the carotid bifurcation until a slight resistance was felt. After 2 h of ischemia, the nylon filament was slowly removed for 24 h of reperfusion. Throughout the procedure, the body temperature was maintained at 36.5 to 37.5°C on a temperature-regulated heating pad. Rats that exhibited abnormal post-operative condition, including heavy neurological deficits such as unconscious and no neurological deficits (neurological score <1), were excluded from the study. In the Sham group, rats were anaesthetized and the carotid artery was exposed, without a filament insert. Rats were randomly divided into four groups (n = 20): Sham, Model, and MPH (0.5 and 1 mg/kg). The MPH doses used were based on the literatures.28,29 Rats in MPH groups were administered 0.5 or 1 mg/kg MPH via i.p. injection for 7 days. Rats in the Sham and Model groups received PBS (vehicle) during the same period. The rats were treated 30 min after ischemia.
Evaluation of neurological deficits
Neurological deficit testing was assessed by an investigator blinded to the experimental groups using a five-point scale (0–4) Bederson’s test (Table 1).30,31
Bederson’s five-point test

Triphenyltetrazolium chloride (TTC) staining
Rat brains were quickly removed and frozen at −20°C for 10 min. 2 mm thick coronal brain sections were sliced and incubated with 2% TTC (Sigma–Aldrich, St. Louis, MO, USA) at 37°C in the dark for 20 min, followed by fixation with 4% paraformaldehyde for 24 h.31,32 Images were photographed using a digital camera. The infarct volume was analyzed by one examiner blinded to the experimental groups using ImageJ 1.43 (National Institutes of Health, Rockville, MD, USA).
Brain water content measurement
Rat brains were quickly removed and samples from the ischemic hemisphere were rapidly weighed to obtain the wet weight (WW), and then dried in an oven at 100°C for 24 h to obtain a dry weight (DW). The brain water content was calculated as: (WW − DW)/WW × 100%.
TUNEL assay
Rat brains were quickly removed and fixed with 4% paraformaldehyde in PBS (pH 7.4) overnight. Brains were then dehydrated using a gradient sucrose solution. Sections (10 µm thick) were made using a freezing microtome. The TUNEL assay was performed according to the manufacturer’s instructions. 3,3′-diaminobenzidine (DAB) was used for the coloration of apoptotic cells. Sections were mounted and the number of TUNEL-positive cells was counted under the BX 60 microscope (Olympus, Tokyo, Japan).31,32
Caspase activity measurement
Rat brains were quickly removed. Brains were homogenized in PBS (pH 7.4) and centrifuged at 3000 rpm for 15 min to remove cellular debris. Supernatants were kept at −80°C and used for further tests. Caspase-3 (Ac-DEVD-Amc, 390/475 nm) and caspase-9 (Ac-LEDH-Afc, 400/505 nm) activities were measured using assay kits according to the manufacturer’s instructions.
Western blot analysis
After treatment, samples were lysed with a lysis buffer (Sigma–Aldrich) containing protease and phosphatase inhibitors to obtain the protein. The protein was quantified using a BCA assay kit (Biyotime, Shanghai, China). Equal amounts of protein (40 μg/lane) were subjected to 4–12% sodium dodecyl sulfate polyacrylamide gel and then transferred onto polyvinylidene difluoride membranes. Membranes were then blocked with 1% bovine serum albumin for 1 h and incubated with the primary antibodies (anti-Bax rabbit mAb ab182733, 1:3000; anti-Bcl-2 rabbit pAb ab194583, 1:1000; anti-AMPK alpha 1 (phospho T183) + AMPK alpha 2 (phospho T172) rabbit mAb ab133448, 1:3000; anti-AMPK alpha 1 + AMPK alpha 2 rabbit mAb ab207442, 1:1000; anti-ACC (phospho S79) rabbit mAb ab68191, 1:5000; anti-ACC rabbit mAb ab45174, 1:1500; anti-β-actin rabbit pAb ab8227, 1:3000) at 4°C overnight. After three washes, membranes were incubated with the secondary antibody (goat anti-rabbit IgG H&L HRP, ab205718, 1:5000) for 2 h at room temperature. Membranes were exposed to ECL substrates (Thermo Scientific, MA, USA), followed by the X-ray film development. 33
Statistical analysis
Data were analyzed with SPSS 19.0 software (SPSS Inc., Chicago, IL, USA). The changes between groups were analyzed by one-way ANOVA followed by LSD post hoc analysis. The neurological score was expressed as the median and analyzed using Kruskal–Wallis one-way analysis of variance on ranks test followed by the post hoc Dunn’s multiple comparison test. A level of p < 0.05 was considered statistically significant.
Results
MPH reduced OGD/R-induced cell damage
Cell viability was examined using the MTT assay and apoptosis was detected by Annexin V/PI staining. Our data showed that MPH treatment alone had no obvious effects on cell viability (Figure 1(a)), which was consistent with findings of the previous study. 23 OGD/R significantly decreased the cell viability and increased the apoptotic rate. However, MPH treatment increased cell viability (Figure 1(b)) and decreased the apoptotic rate (Figure 1(c) and (d)) in a concentration-dependent manner (p < 0.05), thereby reducing OGD/R-induced cell damage.

MPH protected neurons against OGD/R-induced toxicity. Primary neurons were isolated from newborn SD rats. The cell viability was assessed using the MTT assay and cell apoptosis was detected by Annexin V/PI staining. MPH treatment alone had no apparent effects on cell viability (a). MPH at different concentrations (1, 3, and 10 µM) was added 2 h prior to OGD/R. (b) MPH pre-treatment increased cell viability and (c) decreased the apoptotic rate. Representative flow cytometry graphics showed the gating scheme and apoptosis was defined as PI+Annexin V+ (d). Data were expressed as mean ± SD (n = 3). ##p < 0.01 vs. control; *p < 0.05, **p < 0.01 vs. OGD/R group. Experiments were independently performed three times.
MPH exerted protective effects via the AMPK pathway
To explore the MPH mechanism of action on neuroprotective effects in the OGD/R model, the AMPK signaling pathway was investigated. AMPK requires phosphorylation of threonine 172 in the activation loop of the kinase domain for maximal activity. The AMPK activity is represented as the ratio of p-AMPK over total AMPK. 34 Our data showed that MPH treatment increased the protein expression of p-AMPK and p-ACC, while not changing the protein expression of total AMPK and total ACC (Figure 2(a)). To confirm whether AMPK-dependent signaling pathway was the main contributor, the AMPK specific inhibitor was applied prior to MPH exposure. Our results demonstrated that AMPK inhibitor largely abolished MPH-induced neuroprotective effects, evidenced by the decreased cell viability (Figure 2(b)), increased apoptotic rate (Figure 2(c)) and decreased protein expression of p-AMPK as well as p-ACC (Figure 2(d)) (p < 0.05).

MPH exerted protective effects through the AMPK pathway. Primary neurons were isolated from newborn SD rats. The cell viability was assessed using the MTT assay and cell apoptosis was detected by Annexin V/PI staining. MPH was added 2 h prior to OGD/R. To validate the MPH mechanism of action, the specific inhibitor of AMPK Dorsomorphin (10 µM) was added 2 h before MPH treatment. MPH treatment increased p-AMPK and p-ACC protein expression, while not changing total AMPK and total ACC protein expression (a). Pre-treatment with Dorsomorphin (10 µM) largely abolished the neuroprotective effects of MPH, shown by the (b) decreased cell viability, (c) increased apoptotic rate, and (d) decreased protein expression of p-AMPK as well as p-ACC. Data were expressed as mean ± SD (n = 3). ##p < 0.01 vs. control; **p < 0.01 vs. OGD/R group; &p < 0.05 vs. MPH group. Experiments were independently performed three times.
MPH treatment ameliorated brain damage caused by I/R
To further confirm the neuroprotective effects of MPH, the MCAO model was used. Rats in the Sham group did not show any neurological deficits or brain infarct. I/R caused significant neurological damage, evidenced by higher neurological scores, more water content and a larger cerebral infarct size (p < 0.01). However, MPH treatment significantly lowered the neurological scores (Figure 3(a)), water content (Figure 3(b)), and the cerebral infarct size (Figure 3(c)) (p < 0.05), indicating that the brain damage was ameliorated.

MPH treatment significantly ameliorated brain damage caused by I/R. The MCAO method was used to establish I/R injury. Rats were randomly divided into four groups (n = 20): Sham, Model, and MPH (0.5 and 1 mg/kg). In each group, rats were randomly divided into four sub-groups (n = 5) for different sample process. Rats in MPH groups were administered 0.5 or 1 mg/kg MPH via i.p. injection for 7 days. Rats in the Sham and Model groups received PBS during the same period. Rats were treated at 30 min after ischemia. Compared with the model group, MPH treatment significantly decreased (a) the neurological deficit score; (b) brain water content; and (c) cerebral infarct size shown by TTC staining. Data were expressed as median (a, n = 20) or mean ± SD ((b) and (c), n = 5). ##p < 0.01 vs. Sham group; *p < 0.05, **p < 0.01 vs. model group.
MPH treatment decreased apoptosis in brain tissues
TUNEL assay was used to detect the apoptosis in I/R-injured brain tissues. A significant number of TUNEL-positive cells were observed in the Model group, whereas brain sections from the Sham group showed few TUNEL-positive cells. MPH treatment significantly decreased DNA fragmentation (Figure 4(a)) and caspase activities (Figure 4(b)). Moreover, expression of the anti-apoptotic protein Bcl-2 increased, but pro-apoptotic protein Bax expression decreased after MPH treatment (Figure 4(c)) (p < 0.05).
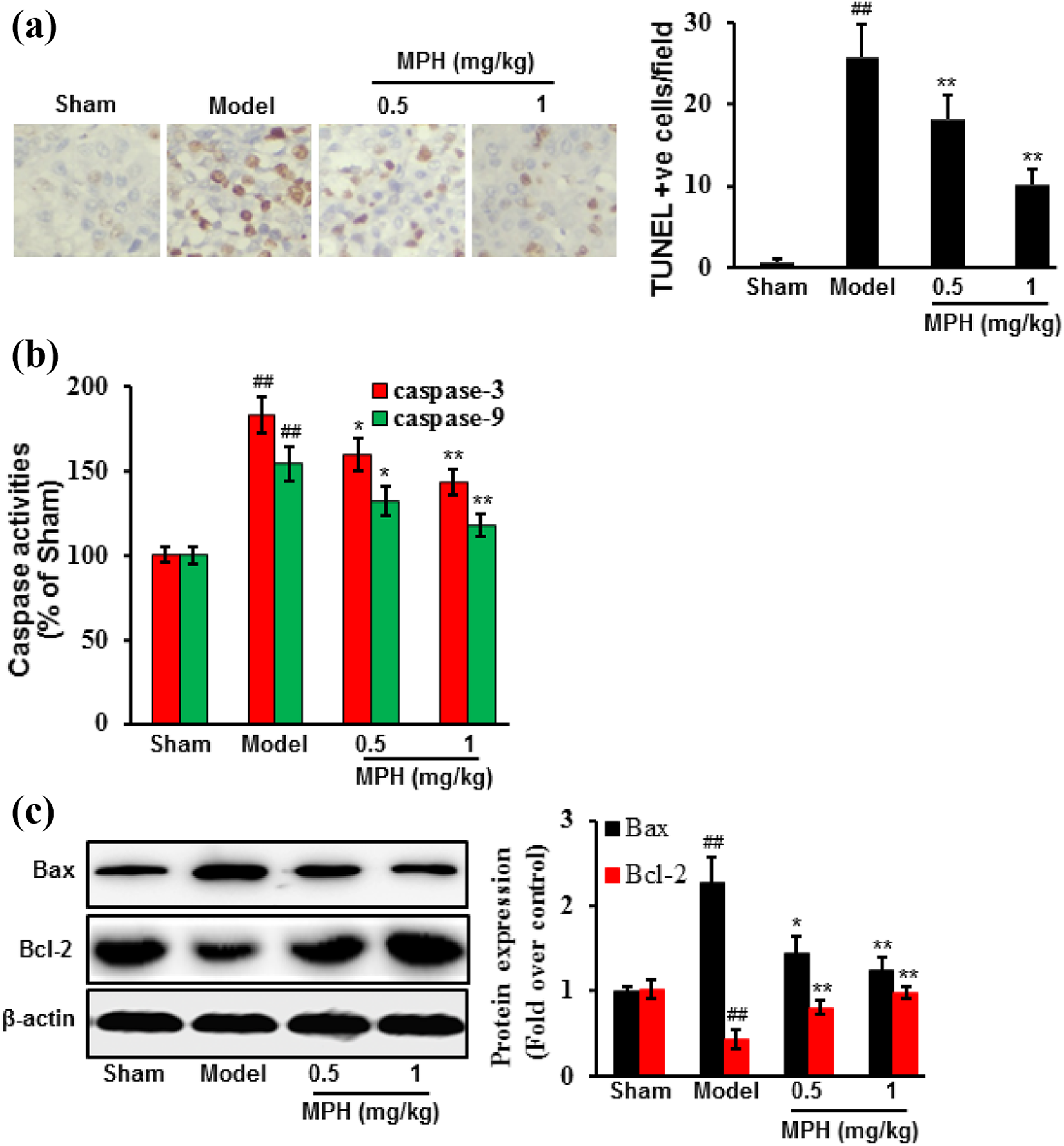
MPH treatment decreased apoptosis in brain tissues induced by I/R. The MCAO method was used to establish I/R injury. Rats were randomly divided into four groups (n = 20): Sham, Model, and MPH (0.5 and 1 mg/kg). In each group, rats were randomly divided into four sub-groups (n = 5) for different sample process. Rats in MPH groups were administered 0.5 or 1 mg/kg MPH via i.p. injection for 7 days. Rats in the Sham and Model groups received PBS during the same period. Rats were treated at 30 min after ischemia. Apoptosis was determined based on: DNA damage shown by the (a) TUNEL assay; (b) caspase activities; and (c) apoptosis-related proteins. Data were expressed as mean ± SD (n = 5). ##p < 0.01 vs. Sham group; *p < 0.05, **p < 0.01 vs. Model group.
MPH treatment increased the protein expression of p-AMPK and p-ACC in brain tissues
To further confirm the MPH mechanism of action, the protein expression of AMPK and ACC was examined. MPH treatment markedly increased the relative protein expression level of p-AMPK and p-ACC (p < 0.05), compared with the Model group, while not changing total AMPK and total ACC protein expression (Figure 5).

MPH treatment increased p-AMPK and p-ACC protein expression in rat brain tissues. The MCAO method was used to establish I/R injury. Rats were randomly divided into four groups (n = 20): Sham, Model, and MPH (0.5 and 1 mg/kg). In each group, rats were randomly divided into 4 sub-groups (n = 5) for different sample process. Rats in MPH groups were administered 0.5 or 1 mg/kg MPH via i.p. injection for 7 days. Rats in the Sham and Model groups received PBS during the same period. Rats were treated 30 min after ischemia. MPH treatment increased the protein expression of p-AMPK and p-ACC, while not changing the protein expression of total AMPK and total ACC. Data were expressed as mean ± SD (n = 5). ##p < 0.01 vs. Sham group; *p < 0.05, **p < 0.01 vs. Model group.
Discussion
Stroke is a leading cause of disability and accounts for 9% of deaths worldwide. Reactive oxygen species (ROS) have been implicated in brain injury after an ischemic stroke. Studies have shown that a rapid increase in ROS production after an acute ischemic stroke overwhelms antioxidant defenses, thus causing further tissue damage. 35 Current treatments to protect the brain against severe stroke damage lack efficacy. A number of agents have been tested and significantly ameliorated the neurological damage associated with focal cerebral I/R injury through inhibiting inflammatory responses and oxidative stress.31,32,36 Unlike these leading compounds, MPH is a first-line stimulant used to treat ADHD. Studies have demonstrated that MPH could ameliorate hypoxia-induced mitochondrial damage in human neuroblastoma SH-SY5Y cells through inhibition of oxidative stress and improve the glutathione status in brains of Wistar rats,22,37 implying its neuroprotective effects. This study investigated protective effects of MPH in OGD/R and MCAO models. Our data indicated that MPH treatment significantly increased cell viability and ameliorated brain damage, which may be associated with the AMPK signaling pathway. Other pathways such as anti-oxidative stress or anti-inflammatory responses may also be involved in the neuroprotective effects of MPH, which will be further investigated in future corollary studies.
During I/R injury, apoptosis plays a pivotal role in brain damage associated with stroke. 38 The Bcl-2 family mainly exerts its pro- or anti-apoptotic activities through the mitochondrial apoptotic pathway. Bax and bid are positive regulators that promote cell death. In contrast, bcl-2 and bcl-xl are negative regulators that prevent cells from undergoing apoptosis. The bcl-2 proteins may facilitate the passage of cytochrome-c or other apoptosis-related factors that trigger the activation of a caspase cascade, resulting in apoptosis.39,40 The caspase family promotes and implements apoptosis in mammalian cells. As an executor, caspase-3 is a key downstream apoptosis protease in the caspase cascade and activates factors of DNA fragmentation, which in turn activate endonucleases to cleave nuclear DNA and cause cell death. 41 In this study, pre-treatment with MPH significantly reduced the loss of cell viability and apoptosis in cultured neurons. Moreover, MPH treatment significantly decreased the activities of caspase-3/9 and DNA fragmentation in brains of MCAO rats. Pro-apoptotic protein Bax expression decreased, but anti-apoptotic protein Bcl-2 expression increased after MPH treatment.
AMPK is activated during events, such as nutrient starvation, which activates a set of energy-producing pathways and inhibits energy-consuming pathways. Thereafter, AMPK acts to balance the levels of ATP consumption and generation via anabolic and catabolic pathways. This role for AMPK is particularly relevant in tissues with a high energy demand and robust ATP consumption.42,43 AMPK is widely but distinctly distributed in different organs, which might explain the susceptibility of different organs to ischemic stimuli. 44 In the brain, ischemia is one of the most important forms of energy deprivation. AMPK is activated by increases in AMP/ATP or ADP/ATP ratios under energy stress. Therefore, AMPK acts as an early sensor of energy deprivation for maintaining metabolic homeostasis.45,46 Neuron apoptosis is usually secondary to ischemic injury and serves as an index of ischemic extent, whereas this process may be ameliorated by AMPK. Cerebral ischemic pre-conditioning can activate AMPK and ameliorate cell apoptosis in the peri-infarct region, evidenced by the reduced percentage of TUNEL-positive cells, suggesting the ability of AMPK to reduce apoptosis in ischemic stroke. 47 Na+/K+-ATPase is a membrane-bound enzyme necessary to maintain neuronal excitability. MPH treatment could increase Na+/K+-ATPase activity in the cerebrum of young and adult rats. 48 Therefore, we hypothesized that MPH exerted its neuroprotective effects in association with AMPK. Our results showed that MPH reduced cell damage through activating the phosphorylation of AMPK, which was confirmed by the addition of an AMPK inhibitor.
Some study limitations should be mentioned. First, it is uncertain whether the data from in vitro and in vivo models accurately reflect molecular changes in human. Second, although clear effects were observed after MPH treatment, its activities need to be confirmed by other animal models. Finally, pharmacokinetic assays are needed to determine the concentration of MPH in different organs and tissues.
In conclusions, this study demonstrated that MPH relieved cerebral I/R injury, at least in part, through activating AMPK. Our data might supply the new insight into the potential of MPH in the treatment of cerebral ischemia.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.