
Abstract
Myocardial ischemia is the main reason for ischemic heart diseases. Antioxidant treatment is considered as a possible approach to prevent myocardial ischemia injury, because oxidative stress is a key factor triggering it. This study was to investigate the protective effects of 3,5-dimethoxy-4-hydroxy myricanol (DHM) against oxidative stress-induced cytotoxicity on H9c2 cells and further explore its mechanisms. The oxidative stress and inflammatory response markers were detected by H2DCFDA fluorescent measurement, enzyme-linked immunosorbent assay (ELISA), real-time PCR and Western blot. Results showed DHM exerted inhibitory effects against H9c2 cell damage. Furthermore, DHM decreased oxidative stress in H9c2 cells through up-regulating protein expression of heme oxygenase-1 (HO-1) and nuclear factor (erythroid-derived 2)-like 2 (Nrf2). Moreover, DHM inhibited inflammatory responses through down-regulating the protein expression of mitogen-activated protein kinases (MAPKs) and nuclear factor kappa B (NF-κB). DHM exerted protective activities against oxidative stress-induced cell damage, at least through decreasing oxidative stress and inhibiting inflammatory responses, indicating that DHM have the potential to be developed as therapeutic agents for the treatment of myocardial ischemia.
Keywords
Introduction
Detailed studies over the past years have proved that persistent myocardial ischemia is the main reason for ischemic heart diseases and sudden death.1,2 The pathophysiological mechanisms of myocardial ischemia are primarily related to pro-inflammatory reactions, which include metabolic disorders, inflammatory responses, and oxidative stress.3,4 Insufficient oxygen supplement will disturb the energy metabolism in cardiomyocytes, causing the damage of cardiomyocytes and inability to support normal function of heart.5,6 Therefore, strategies for exploring the exact molecular mechanism of cardiomyocyte damage under hypoxia environment and searching effective drugs which could prevent cardiomyocytes from damage are of great importance.
Oxidative stress is usually defined as an imbalance between the degradation and generation of reactive oxygen species (ROS) by various antioxidant defense mechanisms. 7 Excessive levels of ROS significantly contribute to the pathogenesis of numerous types of vascular damage. 8 Oxidative stress increases concentrations of intracellular ROS, in turn promoting the release of pro-inflammatory cytokines and cellular apoptosis. Over-production of pro-inflammatory factors resulted in intracellular toxic events, resultantly causing cell death. 9 As one of the regulators of antioxidant cell defense, the nuclear factor erythroid 2-related factor 2 (Nrf2) acts as a protective transcription factor in myocardial ischemia. 10 Hemeoxygenase-1 (HO-1) is one of the antioxidant response element (ARE)-dependent phase II detoxifying enzymes and antioxidants which are regulated by the Nrf2. 11 Activation of Nrf2 induces HO-1 expression, suggesting that Nrf2 is essential for HO-1-mediated cytoprotection against myocardial ischemia. 12
Antioxidant agents from plants have been applied to alleviate the oxidative damage against some disorders, such as retinitis pigmentosa, cataract, Alzheimer’s disease (AD), etc.13–15 Micromelum integerrimum is mainly distributed in China, which has been used for the treatment of cold, trauma, and stomach pain. Chemical investigations of this plant afforded a number of structurally interesting compounds.16–19 As a new compound isolated from the leaves of Micromelum integerrimum, 3,5-dimethoxy-4-hydroxy myricanol (DHM) promoted the proliferation of NIH3T3 cells through up-regulating the protein expression of connective-tissue growth factor. Furthermore, DHM was also reported to ameliorate photoreceptor cell degeneration in Rd10 mice through inhibiting inflammatory responses and endoplasmic reticulum stress.20,21 Therefore, we hypothesized that DHM may protect cardiomyoblast cells against damage caused by oxidative stress and inflammatory responses. In this study, protective effects of DHM were explored against H2O2-mediated cell damage on H9c2 cells. Our results indicated that DHM significantly inhibited apoptosis and increased cell viability for H9c2 cells under oxidative stress.
Materials and methods
Cell lines, reagents and chemicals
H9c2 and HL-1 cell lines were purchased from Shanghai Cell bank (Shanghai, China). Fetal bovine serum (FBS) and RPMI1640 medium were purchased from Gibco BRL (Waltham, MA, USA). 1-Methyl-4-phenylpyridinium (MPP+), and 3-(4,5-dimethylthiazol)-2,5-diphenyltetrazolium-bromid (MTT) were from Sigma-Aldrich (St. Louis, MO, USA). Glutathione (GSH), superoxide dismutase (SOD), malondialdehyde (MDA), interleukin-1β (IL-1β), interleukin-6 (IL-6) and tumor necrosis factor alpha (TNFα) assay kits were purchased from Jiancheng Biological Engineering (Nanjing, China). Cytochrome-c assay kit was from R&D Systems (Minneapolis, MN, USA). Caspase-3 and Caspase-9 activity assay kits were from My BioSource (San Diego, CA, USA). Annexin V apoptosis assay kit was purchased from ThermoFisher (Waltham, MA, USA). 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) and 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) were from Molecular Probes (Eugene, OR, USA). Real-time PCR reagents were from ThermoFisher (Waltham, MA, USA). All solvents and chemicals used in this study were from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China) as analytical grade. 30% (w/w) H2O2 (9.8 M) was freshly diluted with Milli-Q water to 300 mM before treatment. DHM (purity >98%) was purchased from WuXi AppTec Company (Shanghai, China). DHM were dissolved in DMSO for the in vitro studies.
Cell culture and treatment
Cells were cultured in RPMI1640 medium supplemented with 10% FBS and 1% streptomycin/penicillin at 37°C. Cells were treated with DHM at 0.1, 0.5, 2.5 µM for 6 h, and followed by H2O2 at 300 µM or MPP+ at 500 µM for another 24 h. Concentrations of H2O2 and MPP+ were chosen based on our preliminary studies and literatures.22–24 Cells were treated with DMSO as the negative control. Cell number was counted by the Vi-Cell Cell Viability Analyzer (Beckman Coulter, Brea, CA, USA).
Cell viability assay
The cell viability was assessed by MTT assay. After treatment with DHM at 0.1, 0.5, 2.5 µM for 6 h, and followed by H2O2 at 300 µM or MPP+ at 500 µM for another 24 h, cells were seeded into 96-well plates at 50,000 cells per well. Cells were then incubated with 0.5 mg/mL MTT at 37°C for 4 h. The formazan was dissolved in 100 µL DMSO and plates were kept at room temperature for 2 h. The absorbance was determined at 570 nm with a microplate reader.
Mitochondrial membrane potential (MMP) measurement
After treatment with DHM at 0.1, 0.5, 2.5 µM for 6 h, and followed by H2O2 at 300 µM for another 24 h, cells were seeded into 96-well plates at 30,000 cells per well. 10 µL of 200 µM JC-1 (2 µM final concentration) was added and incubated with cells at 37°C for 15 min in the dark. After washing twice by adding 200 µL PBS, cells were re-suspended in 100 µL PBS. The fluorescence intensity was determined by the microplate reader (TECAN Polarion) with 488 nm excitation and 530/590 nm emission, respectively.
Cytochrome-c measurement
After treatment with DHM at 0.1, 0.5, 2.5 µM for 6 h, and followed by H2O2 at 300 µM for another 24 h, H9c2 cells were re-suspended with 1 mL of extraction buffer. Cells were then homogenized in an ice-cold Dounce tissue grinder (Catalog No. 1998 -1, BioVision). The fraction of cytoplasmic extract was used for cytochrome-c measurement with the quantikine ELISA kit (Cat. No. DCTC0; R&D Systems, Inc.). Briefly, add 100 µL of calibrator diluent to each well of 96-well microplate; add 100 µL of standard, control, or sample to each well, cover with a plate sealer, and incubate at room temperature for 2 h; aspirate each well and wash, repeating 3 times; add 200 µL of conjugate to each well, cover with a new plate sealer, and incubate at room temperature for 2 h; aspirate and wash 4 times; add 200 µL substrate solution to each well and incubate at room temperature for 30 min; add 50 µL of stop solution to each well, and read at 450 nm within 30 min.
Caspase activity measurement
After treatment with DHM at 0.1, 0.5, 2.5 µM for 6 h, and followed by H2O2 at 300 µM for another 24 h, cells were seeded into 96-well plates at 40,000 cells per well. Activities of caspase-3 (Ac-DEVD-Amc, Catalog No.: MBS261814, My BioSource) and caspase-9 (Ac-LEDH-Afc, Catalog No.: MBS264306, My BioSource) were measured by the assay kits following instructions of the manufacturer. Briefly, add 50 µL of 2× reaction buffer to each well; add 5 µL of substrates (200 µM final concentration); cover the plate with a plate sealer or lid; gently mix contents of wells using a plate shaker at 300 rpm for 30 s; incubate at 37°C for 2 h; measure the OD values with a microplate reader (caspase-3, 390/475 nm; caspase-9 400/505 nm).
Apoptosis analysis
After treatment with DHM at 0.1, 0.5, 2.5 µM for 6 h, and followed by H2O2 at 300 µM for another 24 h, 3 × 106 H9c2 cells were collected and washed with ice-cold PBS. Cells were re-suspended in 500 µL binding buffer and incubated with 5 µL Annexin V-FITC/PI (Catalog No.: 88-8005-74, ThermoFisher) for 10 min in the dark. Apoptosis was detected with FACScan flow cytometer (Becton Dickinson). Data were analyzed by FlowJo V10.
ROS measurement
H2DCFDA was used to measure ROS production in H9c2 cells. After treatment with DHM at 0.1, 0.5, 2.5 µM for 6 h, and followed by H2O2 at 300 µM for another 24 h, 1 × 105 H9c2 cells in 200 µL PBS were added in 96-well plate and incubated with H2DCFDA (10 µM final concentration) for 20 min at 37°C. After washing twice with PBS, ROS were measured using a fluorescence microplate reader.
SOD activity, MDA and GSH level
After treatment with DHM at 0.1, 0.5, 2.5 µM for 6 h, and followed by H2O2 at 300 µM for another 24 h, H9c2 cells were collected and homogenized. The supernatant was used to measure SOD activity (Catalog No.: A001-3-2), GSH (Catalog No.: A006-2-1) and MDA (Catalog No.: A003-1-2) levels with assay kits following manufacturers’ instructions (Jiancheng Biological Engineering, Nanjing, China).
NO and cytokine measurement
After treatment with DHM at 0.1, 0.5, 2.5 µM for 6 h, and followed by H2O2 at 300 µM for another 24 h, H9c2 cells were collected and homogenized. NO was measured by Griess assay kit (Catalog No.: G-7921, Molecular Probes, Eugene, OR, USA). Levels of IL-1β (Catalog No.: H002), IL-6 (Catalog No.: H007) and TNFα (Catalog No.: H052) were measured by assay kits following manufacturers’ instructions (Jiancheng Biological Engineering, Nanjing, China).
Real-time PCR
Total RNA was extracted with RNeasy reagents and mRNA was transcribed into cDNA using iScriptTM Reverse Transcription Supermix. Quantitative PCR was run on StepOne systems (Thermo Fisher) using SYBRTM Green Master Mix with the 2−ΔΔCt method. β-actin was used as the internal reference. The primer sequences are: for iNOS, forward: ggctagcattccaaaagctg; reverse: cttagggctgcctttctgtg, species: Rat, GenBank: AJ230461.1; for IL-1β, forward: aggcttccttgtgcaagtgt; reverse: tgagtgacactgccttcctg, species: Rat, GenBank: M98820.1; for IL-6, forward: ccggagaggagacttcacag; reverse: acagtgcatcatcgctgttc, species: Rat, GenBank: M26744.1; for TNFα, forward: agatgtggaactggcagagg; reverse: cccatttgggaacttctcct, species: Rat, GenBank: X66539.1; for β-actin, forward: agccatgtacgtagccatcc; reverse: ctctcagctgtggtggtgaa, species: Rat, GenBank: FQ227027.1.
Western blot analysis
After treatment with DHM at 0.1, 0.5, 2.5 µM for 6 h, and followed by H2O2 at 300 µM for another 24 h, H9c2 cells were lysed with RIPA buffer, supplemented with protease inhibitors, phosphatase inhibitors, 1 mM EDTA and 1 mM PMSF. Protein concentration was determined with the BCA assay kit. After boiling for 5 min, 40 µg sample protein with 4× loading dye was loaded in each well of 4–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis. After running, the protein was transferred onto polyvinylidene difluoride membranes. The membrane was blocked with 1% bovine serum albumin at room temperature for 1 h and then incubated with primary antibodies (anti-Cleaved Caspase-3 rabbit pAb ab2302, 1:500; anti-Cleaved Caspase-9 rabbit pAb orb159342, 1:1000; anti-Bax rabbit pAb ab53154, 1:1000; anti-Bcl-2 rabbit pAb ab59348, 1:1000; anti-NADPH oxidase 4 rabbit pAb ab154244, 1:1500; anti-HO-1 rabbit pAb ab13243, 1:1500; anti-Nrf2 rabbit pAb ab92946, 1:1000; anti-JNK1 (phosphor T183) rabbit pAb ab47337, 1:500; anti-P38 (phosphor Y182) rabbit pAb ab47363, 1:500; anti-P65 (phospho S529) rabbit pAb ab97726, 1:1000; anti-β-actin rabbit pAb ab227387, 1:5000) at 4°C overnight. The membrane was washed three times and incubated with the secondary antibody (goat anti-rabbit IgG H&L (HRP), ab6721, 1:5000) at room temperature for 1 h. The chemiluminescent signals were detected by the ChemiDoc MP Imaging System (Bio-Rad, Hercules, CA, USA).
Statistical analysis
Data were presented as mean ± standard deviation (SD) from three independent experiments and analyzed with SPSS12.0. Measurement data between two groups were compared using the t-test; measurement data among multiple groups were compared using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. The minimal level of significance was p < 0.05.
Results
DHM exerted protective effects on H9c2 cells
Compared with the negative control, no significant change was observed by MTT assay and cell count, suggesting DHM treatment at 2.5 µM didn’t cause H9c2 cell damage (Figure 1). Treating H9c2 cells with H2O2 alone resulted in apparent cell damage, whereas DHM treatment increased cell viability. To confirm mechanisms of DHM, the other oxidative stress model, MPP+ model, was used in this study. Our data showed that treatment with DHM protected H9c2 cells against damage caused by MPP+ (Figure 1). Moreover, another cell line HL-1 was used to confirm protective effects of DHM. Results demonstrated that DHM could also protect HL-1 cells against the oxidative damage (Figure 2).
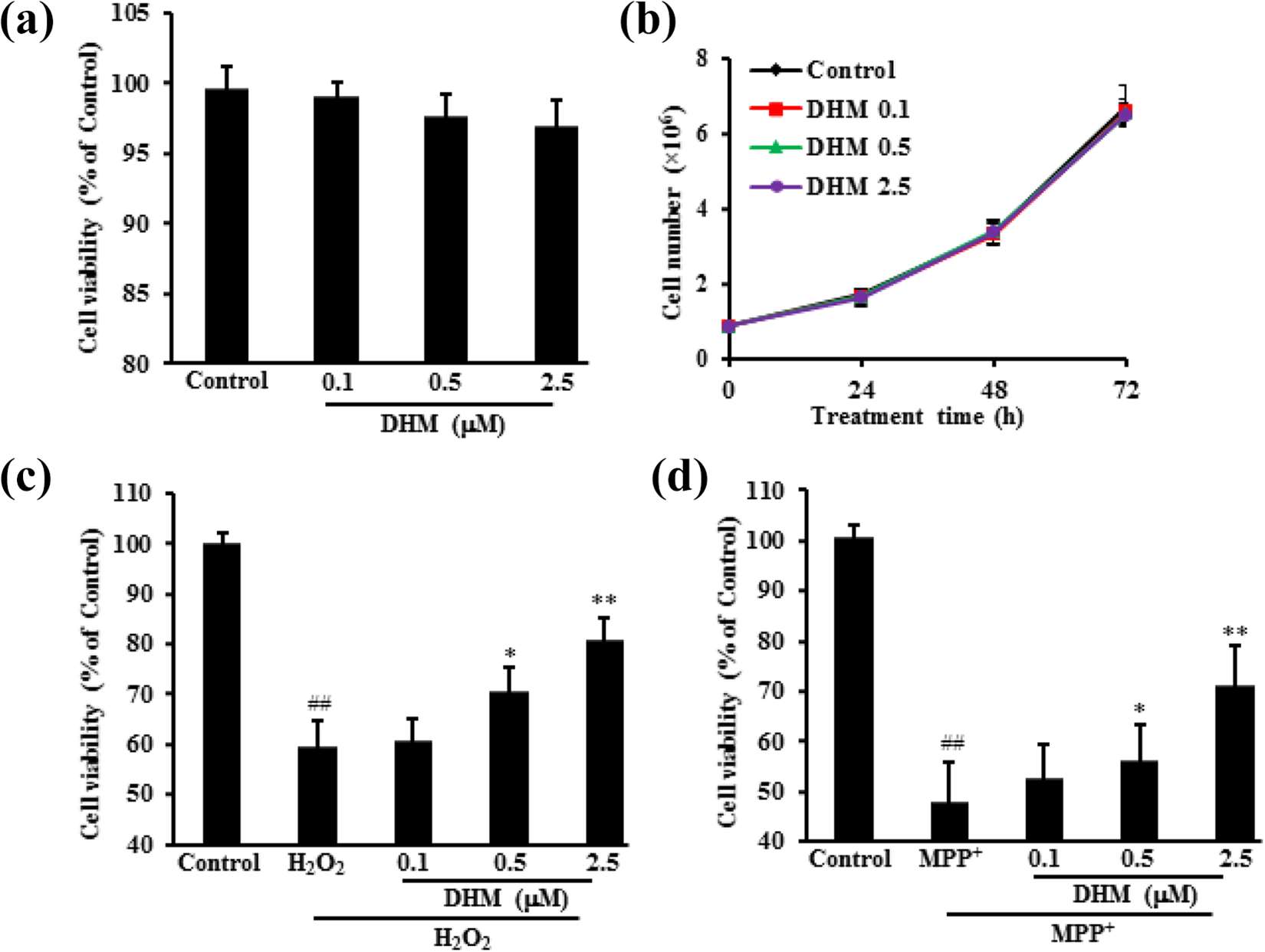
Effects of DHM treatment on H9c2 cell viability and its protective effects. After treating H9c2 cells with DHM at 0.1, 0.5, 2.5 µM for 72 h, cell viability was detected by MTT assay and the cell number was also counted. H9c2 cells were firstly treated with DHM at 0.1, 0.5, 2.5 µM for 6 h and then with H2O2 or MPP+ for another 24 h. H9c2 cells were collected to measure the cytotoxicity. DHM had no apparent effects on H9c2 cell proliferation measured by MTT assay (a) and cell count (b). DHM protected H9c2 cells against damage caused by H2O2 (c) or MPP+ (d). Data were expressed as mean ± SD. ##p < 0.01 vs. Control; *p < 0.05, **p < 0.01 vs. H2O2 or MPP+ alone.
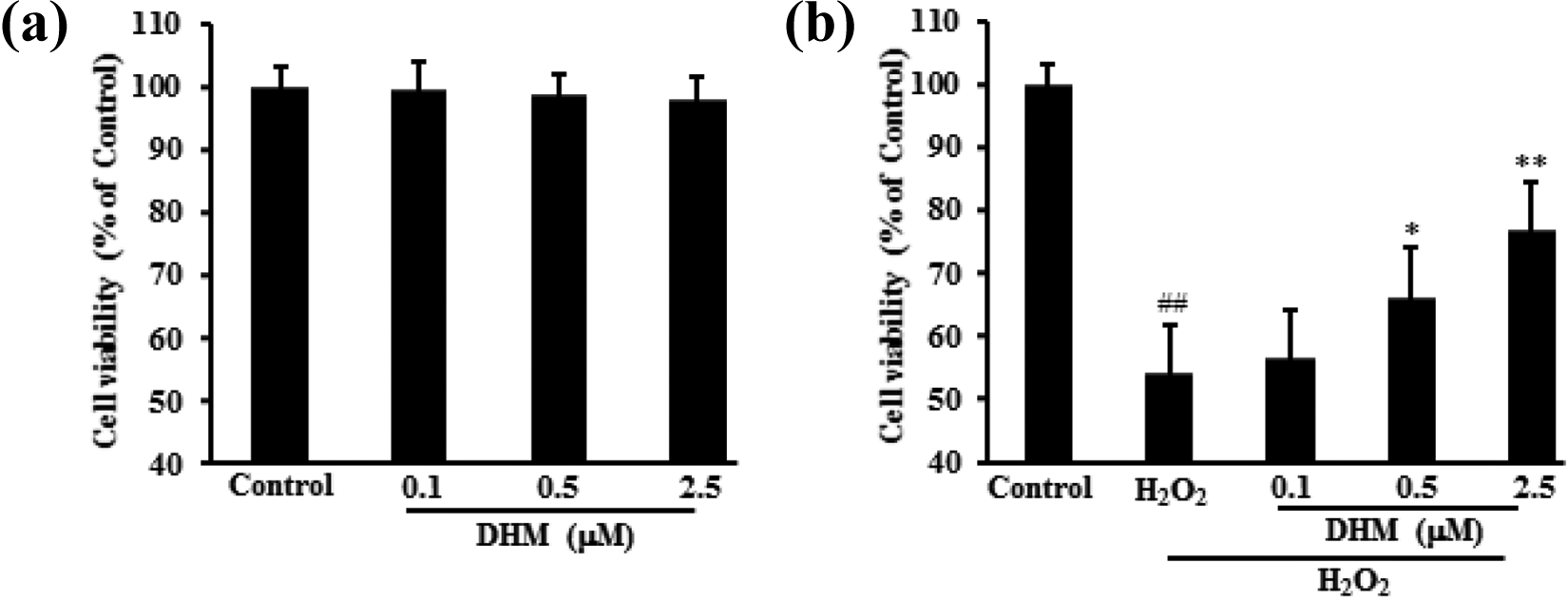
Effects of DHM treatment on HL-1 cell viability and its protective effects. After treating HL-1 cells with DHM at 0.1, 0.5, 2.5 µM for 72 h, cell viability was detected by MTT assay. HL-1 cells were firstly treated with DHM at 0.1, 0.5, 2.5 µM for 6 h and then with H2O2 for another 24 h. HL-1 cells were collected to measure the cytotoxicity. DHM had no apparent effects on HL-1 cell proliferation measured by MTT assay (a). DHM protected HL-1 cells against damage caused by H2O2 (b). Data were expressed as mean ± SD. ##p < 0.01 vs. Control; *p < 0.05, **p < 0.01 vs. H2O2 alone.
DHM decreased H9c2 cell apoptosis caused by H2O2
After treatment, apoptosis-related biomarkers were detected. Compared with the H2O2 group, DHM treatment increased MMP; decreased cytochrome-c release as well as Caspase-3/9 activities in H9c2 cells; and decreased apoptotic rate. Pro-apoptotic protein cleaved Caspase-3/9 and Bax expression decreased, but anti-apoptotic protein Bcl-2 expression significantly increased after DHM treatment (Figure 3).
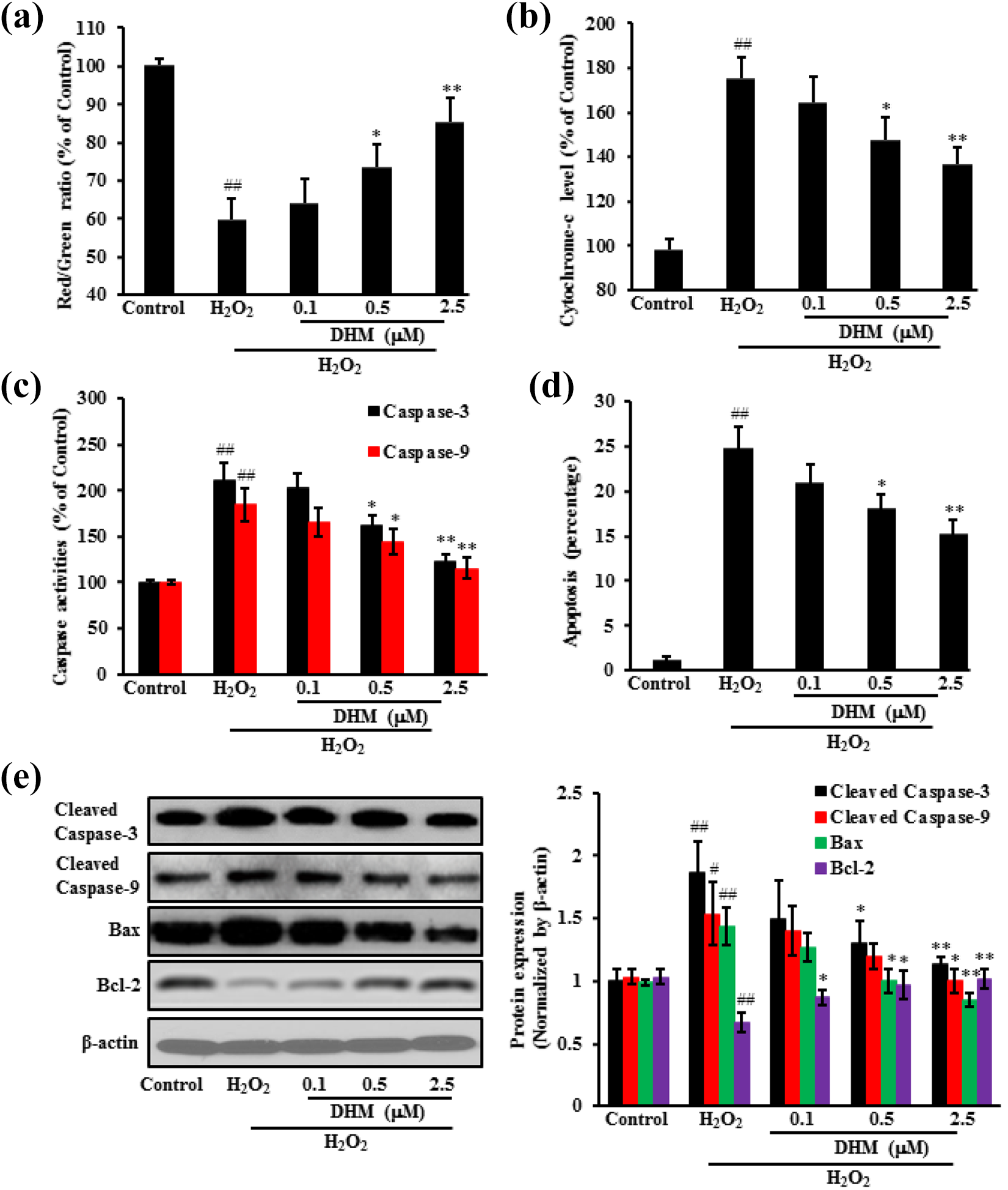
DHM protected H9c2 cells against H2O2-induced cell apoptosis. After treating H9c2 cells with DHM at 0.1, 0.5, 2.5 µM for 6 h and then with H2O2 for another 24 h, cells were collected to measure mitochondrial membrane potential (a); cytochrome-c release (b); caspase activities (c); and apoptosis detected by Annexin V/PI staining (d). Cleaved Caspase-3/9, Bax and Bcl-2 proteins were also detected by western blot (e). Data were expressed as mean ± SD. ##p < 0.01 vs. Control; *p < 0.05, **p < 0.01 vs. H2O2 alone.
DHM decreased the oxidative stress in H9c2 cells
H2O2 significantly increased the oxidative stress. However, DHM treatment significantly decreased the MDA level and ROS production, but increased SOD activities and GSH levels. Moreover, DHM treatment down-regulated protein expression of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 and up-regulated protein expression of HO-1 as well as Nrf2. These data demonstrated that DHM effectively alleviated the oxidative stress caused by H2O2 (Figure 4).
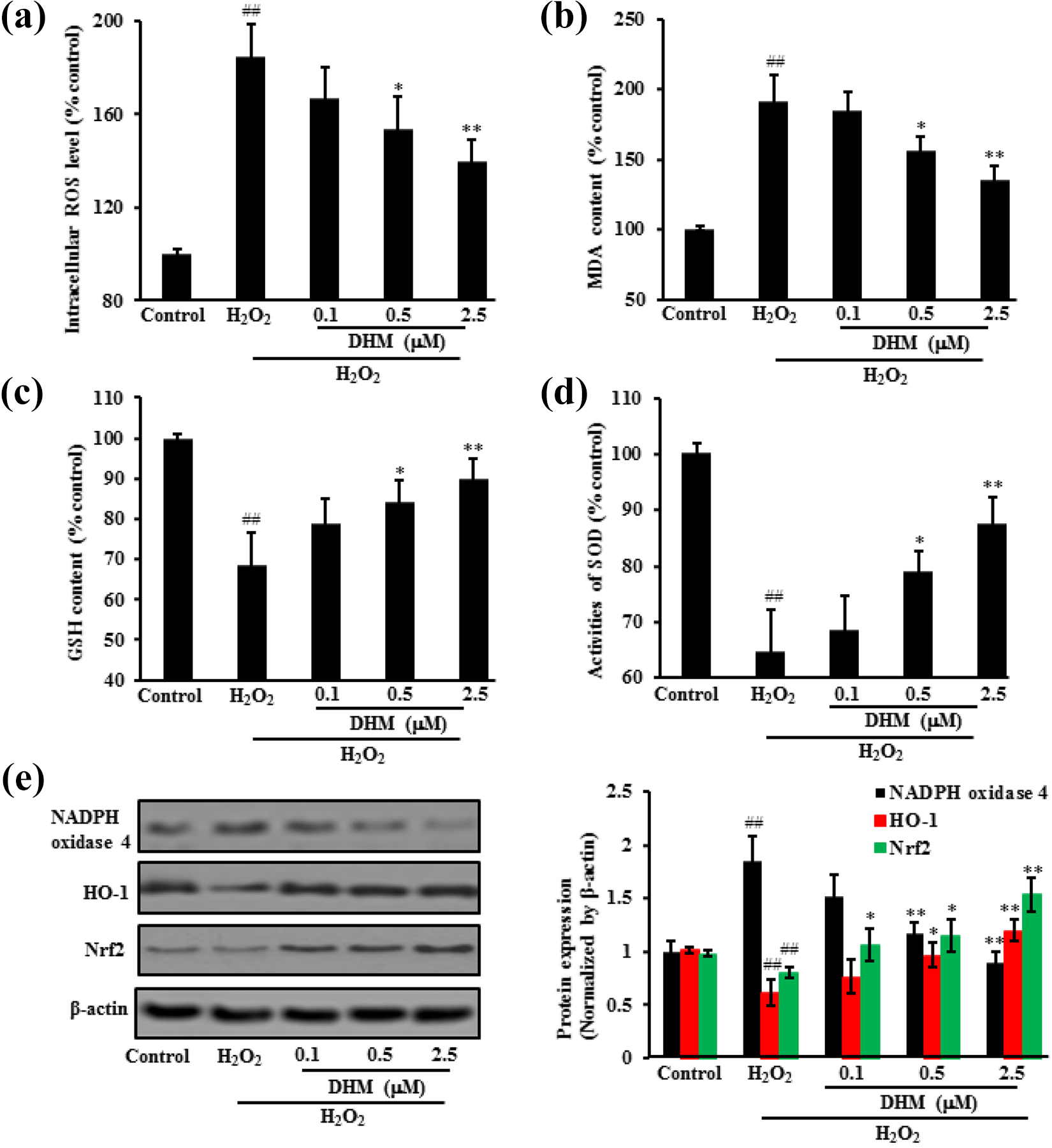
DHM decreased oxidative stress caused by H2O2 in H9c2 cells. After treating H9c2 cells with DHM at 0.1, 0.5, 2.5 µM for 6 h and then with H2O2 for another 24 h, the oxidative stress markers were measured by: ROS level (a); MDA content (b); GSH level (c), SOD activity (d) and oxidative stress-related protein expression (e). Data were expressed as mean ± SD. ##p < 0.01 vs. Control; *p < 0.05, **p < 0.01 vs. H2O2 alone.
DHM inhibited inflammatory responses in H9c2 cells
DHM treatment exerted significant inhibitory effects on the production of NO, IL-1β, IL-6 and TNFα in H2O2-treated H9c2 cells. Gene expression of iNOS, IL-1β, IL-6 and TNFα also increased following exposure to H2O2, but significantly decreased after DHM treatment. Furthermore, H2O2 increased protein expression of p-JNK, p-P38 and p-P65, but DHM treatment significantly decreased expression levels of these proteins (Figure 5).

DHM inhibited inflammatory responses induced by H2O2 in H9c2 cells. After treating H9c2 cells with DHM at 0.1, 0.5, 2.5 µM for 6 h and then with H2O2 for another 24 h, inflammatory responses were measured by: NO (a); IL-1β (b); IL-6 (c); TNFα (d); and gene as well as protein expression (e, f). Data were expressed as mean ± SD. ##p < 0.01 vs. control; *p < 0.05, **p < 0.01 vs. H2O2 alone.
Discussion
Oxidative stress and inflammatory responses have been confirmed to be associated with the pathophysiology of myocardial ischemia. 25 Thereafter, administration of exogenous antioxidant agents provides a strategy to attenuate the redox imbalance and improve heart function. 26 DHM has been reported to ameliorate photoreceptor cell degeneration in Rd10 mice through inhibiting inflammatory responses. 21 The present study further demonstrated that DHM, as a novel antioxidant, induced Nrf2 which appeared to be involved in the intracellular defense mechanisms against H2O2-mediated oxidative stress. Moreover, DHM suppressed H2O2-mediated over-activation of upstream signaling molecules, including NF-κB and MAPKs, which were involved in inflammatory responses. These results revealed that DHM attenuated the toxicity of H2O2 and possessed anti-inflammatory effects to protect cardiomyoblast cells.
Mitochondria play an important role during the process of cell death regulation. Decrease of mitochondrial membrane potential causes mitochondrial fragmentation, resultantly generating excessive amounts of ROS, causing mitochondrial structure and function damage, eventually interrupting the cellular ATP supply and initiating the apoptosis.27,28 Mitochondria modulated cell viability through ROS-mediated oxidative injury and the high temperature requirement protein A2 (HtrA2/Omi) liberation-induced caspase activation.29,30 As an executor, caspase-3 activates factors of DNA fragmentation, which in turn activate endonucleases to cleave nuclear DNA to cause cell death. 31 In the present study, H2O2 caused H9c2 cell damage, evidenced by the decrease of MMP, more cytochrome-c release, and elevated activities of caspase-3/9. Expression of anti-apoptotic protein Bcl-2 down-regulated, but expression of pro-apoptotic protein Bax, cleaved Caspase-3/9 up-regulated. However, these effects were largely reversed by DHM treatment.
Over-production of ROS can result in oxidative stress, which causes different cell death patterns, including autophagy, apoptosis and necrosis.32,33 Oxidative stress has been considered as one of major causes of cell damage in many disorders.34,35 Therefore, it is reasonable to use external antioxidants to restore the balance between intracellular oxidative and anti-oxidative systems. The transcription factor Nrf2 is a master regulator of detoxificant and antioxidant genes with cytoprotective function. 36 Since inactivation of Nrf2 is necessary for the complete execution of apoptosis in the presence of cellular damage caused by oxidative stress, constant activation of Nrf2 may protect cells from apoptosis, through binding with antioxidant response element to up-regulate protein expression of cytoprotective targets, such as antioxidant proteins, phase II detoxifying enzymes, and molecular proteasome/chaperones.37,38 In the present study, DHM-induced Nrf2 activation was observed, which further increased expression of antioxidants to restore oxidative homeostasis, demonstrated by up-regulated protein expression of HO-1, down-regulated protein expression of NADPH oxidase 4, reduced levels of ROS as well as MDA, and increased GSH level as well as SOD activity.
The production of inflammatory mediators participates in immune responses to many inflammatory stimuli and is involved in the progress of inflammatory diseases. 39 DHM exerted anti-inflammatory effects by inhibition of inflammatory response inducers, including pro-inflammatory mediator NO, and pro-inflammatory cytokines IL-1β, IL-6 as well as TNFα in the H2O2-stimulated H9c2 cells. These cytokines stimulate the production of oxidants with subsequent peroxidative damage to various macromolecules. 40 Activation of NF-κB (P65) and MAPKs leads to transcription factor binding to the promoter regions of pro-inflammatory cytokines, thereby enabling transduction of extracellular signals into cellular reactions. 41 DHM decreased expression of several MAPKs induced by H2O2 stimulation, including p-JNK and p-P38. In addition, DHM reduced expression of p-NF-κB (p-P65) in response to H2O2.
In conclusions, our data demonstrated that DHM significantly inhibited H2O2-induced oxidative stress and inflammatory responses in the in vitro model of H9c2 cells. The in vivo animal models will be helpful to confirm the therapeutic efficacy of DHM and further explore its exact mechanisms.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.