
Abstract
The aim of this study was to examine the effects of lipid emulsions on carnitine palmitoyltransferase I (CPT-I), carnitine acylcarnitine translocase (CACT), carnitine palmitoyltransferase II (CPT-II), and the mitochondrial dysfunctions induced by toxic doses of local anesthetics in H9c2 rat cardiomyoblasts. The effects of local anesthetics and lipid emulsions on the activities of CPT-I, CACT, and CPT-II, and concentrations of local anesthetics were examined. The effects of lipid emulsions, N-acetyl-L-cysteine (NAC), and mitotempo on the bupivacaine-induced changes in cell viability, reactive oxygen species (ROS) levels, mitochondrial membrane potential (MMP), and intracellular calcium levels were examined. CACT, without significantly altering CPT-I and CPT-II, was inhibited by toxic concentration of local anesthetics. The levobupivacaine- and bupivacaine-induced inhibition of CACT was attenuated by all concentrations of lipid emulsion, whereas the ropivacaine-induced inhibition of CACT was attenuated by medium and high concentrations of lipid emulsion. The concentration of levobupivacaine was slightly attenuated by lipid emulsion. The bupivacaine-induced increase of ROS and calcium and the bupivacaine-induced decrease of MMP were attenuated by ROS scavengers NAC and mitotempo, and the lipid emulsion. Collectively, these results suggested that the lipid emulsion attenuated the levobupivacaine-induced inhibition of CACT, probably through the lipid emulsion-mediated sequestration of levobupivacaine.
Keywords
Introduction
Lipid emulsions are used to treat systemic toxicity caused by local anesthetics. 1 The suggested underlying mechanisms of action include lipid shuttling, inotropic effects, fatty acid supplementation, attenuation of mitochondrial dysfunction, glycogen synthase kinase-3β phosphorylation, inhibition of nitric oxide release, and calcium influx. 1 Lipid shuttling, a widely accepted underlying mechanism, refers to the absorption of highly lipid soluble drugs (for example: bupivacaine; log[octanol/water partition coefficient]: 3.41) by lipid emulsions in the heart, and their subsequent transportation to the liver and adipose tissue, resulting in their enhanced redistribution. 1 In addition, lipid emulsions have been reported to reverse the vasodilation induced by toxic doses of aminoamide local anesthetics, including levobupivacaine, bupivacaine, ropivacaine, lidocaine, and mepivacaine, which seem to function in a lipid solubility-dependent pattern of local anesthetics.2,3 Fatty acid oxidation of lipid emulsions is known to be involved in the recovery of bupivacaine-induced cardiac depression. 4 In addition, pretreatment with adenosine triphosphate (ATP) was reported to nearly restore the bupivacaine-induced decreased myocardial contractility. 5 As the supply of fatty acids, as a major energy source, produces ATP in cardiac mitochondria via beta-oxidation, it was suggested that the supply of fatty acids might partially contribute to the lipid emulsion-mediated reversal of cardiac toxicity induced by toxic doses of bupivacaine.1,4,5 Carnitine palmitoyltransferase I (CPT-I), carnitine acylcarnitine translocase (CACT), and carnitine palmitoyltransferase II (CPT-II) are involved in transporting long chain fatty acids from the cytoplasm to the mitochondria matrix, which is preferentially used as an energy source in the heart. 6 Bupivacaine has been shown to inhibit CACT, thus leading to the inhibition of the transportation of long-chain fatty acids and carnitine shuttling. 7 However, the effects of lipid emulsions on the activities of CPT-I, CACT, and CPT-II following toxic doses of local anesthetics remain unknown.
The production of mitochondrial reactive oxygen species (ROS) induces the depolarization of the mitochondrial membrane and increases the levels of calcium. 8 The highly lipid soluble local anesthetic bupivacaine has been reported to inhibit ATP synthesis in cardiac mitochondria, leading to bupivacaine-induced myocardial depression via the direct inhibition of complex I in the electron transport chain.9,10 In contrast, lipid emulsions were shown to attenuate doxorubicin-induced cardiac toxicity via the inhibition of oxidative stress. 11 In addition, lipid emulsions have been reported to inhibit bupivacaine-induced apoptosis, potentially through the reduction in cellular oxidative stress. 12
Based on these findings, we tested the biological hypothesis that lipid emulsions would attenuate bupivacaine-induced cardiotoxicity via the reversal of the decreased activity of CACT and inhibition of mitochondrial dysfunction.1,4,5,7–12 The aim of this in vitro study was to examine the effects of lipid emulsions (Intralipids) on the activities of CPT-I, CACT, and CPT-II following the administration of toxic doses of local anesthetics in H9c2 rat cardiomyoblasts. Furthermore, we aimed to examine the effects of lipid emulsions on the bupivacaine-induced mitochondrial dysfunction in H9c2 rat cardiomyoblasts.
Materials and methods
Cell culture
H9c2 cardiomyoblasts, obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA), were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco), 100 units/mL penicillin, and 100 μg/mL streptomycin (Gibco). Cells were maintained in a humidified incubator maintained at 5% CO2 and 37°C. For experiments, cells were starved in serum-free medium for 16 h before drug treatment.
Determination of CPT-I, CPT-II, and CACT Activities
Enzyme-linked immunosorbent assay (ELISA) kits were employed to evaluate the activities of CPT-I and CPT-II (MyBioSource, San Diego, CA, USA) and CACT (Elabscience, Houston, Texas, USA). H9c2 cells were treated with local anesthetics (7 × 10-4 M levobupivacaine and bupivacaine, 10-3 M ropivacaine, and 10-2 M mepivacaine), alone or combined with a lipid emulsion (0.05, 0.1 and 0.2%).13–16 H9c2 cells were treated with levobupivacaine, ropivacaine, and mepivacaine for 3 or 10 h to measure CPT-I and CPT-II activity. H9c2 cells were treated with local anesthetics alone (levobupivacaine, bupivacaine, ropivacaine, and mepivacaine) for 3 h, lipid emulsion for 1 h followed by local anesthetics for 3 h, or lipid emulsion alone for 4 h, to measure CACT activity. The culture medium was collected and centrifuged at 10 000 × g and 4°C for 20 min. Subsequently, the activities of CPT-I, CPT-II, and CACT in the supernatant were measured using the respective ELISA kits according to the manufacturer’s instructions. Absorbance was read at 450 nm using a VersaMax® microplate reader (Molecular Devices, Sunnyvale, CA, USA). The activities of CPT-I, CPT-II and CACT were determined by comparing the optical density values of the samples to a standard curve.
Effect of a lipid emulsion on the concentrations of local anesthetics in krebs solution
Toxic concentrations of local anesthetics (7 × 10-4 M levobupivacaine, 10-3 M ropivacaine, and 10-2 M mepivacaine) were emulsified with a lipid emulsion (Intralipid: 0, 0.05, 0.1, and 0.2%) in Krebs solution using a rotator for 30 min, as previously described.13,15–17 After emulsification, emulsified samples were centrifuged at 75 000 × g for 40 min to assess the release of local anesthetic from samples. The concentration of each local anesthetic in the aqueous phase, which is equivalent to the centrifuged aqueous extract, was measured with ultraperformance liquid chromatography-quadrupole time-of-flight mass spectrometry (UPLC-Q-TOF MS; Waters, Milford, MA, USA) using an Acquity UPLC BEH C18 column (100 × 2.1 mm, 1.7 μm; Waters). The aqueous layer was injected into the column equilibrated with water/acetonitrile (99:1) containing 0.1% formic acid (FA) and eluted with a linear gradient (1–100%) of acetonitrile, containing 0.1% FA, at a flow rate of 0.35 mL/min for 5 min. The eluted local anesthetic was analyzed via Q-TOF MS with multiple reactions monitoring in a positive electrospray ionization mode. The sampling and capillary cone voltages were set at 30 kV and 3 V, respectively. The source and desolvation temperatures were set at 400 and 100°C, respectively, while the desolvation flow rate was 800 L/h. The eluted local anesthetic was monitored by the product and precursor ions of m/z 294.08 and 409.14, respectively. LockSpray with leucine-enkephalin ([M+H] = 556.2771 Da) was used to ensure the reproducibility and accuracy of all analyses. All mass data were collected and analyzed using UIFI 1.8.2 (Waters).
Cell viability assay
Cell viability was determined using the Cell Counting Kit eight assay (CCK-8) (Dojindo Molecular Technologies, Kumamoto, Japan), as previously described. 17 In brief, H9c2 cells were seeded into 24-well plates at a density of 2 × 104 cells/well. H9c2 cells were then treated with bupivacaine alone (10-3 M) for 36 h, lipid emulsion (0.1, 0.2, and 0.3%) for 1 h followed by bupivacaine (10-3 M) for 36 h, lipid emulsion (0.1, 0.2, and 0.3%) alone for 37 h, the inhibitor (oxfenicine [2 × 10-3 M], LY-294002 [3 × 10-6 M], and SB216763 [5 × 10-6 M]) for 1 h followed by lipid emulsion (1%) for 1 h and post-treatment with bupivacaine (10-3 M) for 36 h, or the inhibitor and dimethyl sulfoxide (DMSO, 0.11%) alone for 38 h. In addition, H9c2 cells were treated with bupivacaine (2 × 10-3 M) for 12 h, N-acetyl-L-cysteine (NAC, 3 × 10-2 M) for 1 h followed by bupivacaine (2 × 10-3 M) for 12 h, or NAC (3 × 10-2 M) alone for 13 h. In addition, after treatment, 500 μL DMEM, containing a 10% CCK-8 solution, was added to each well and cells were incubated for an additional 3 h at 37°C in a 5% CO2 atmosphere. The optical density of each well was measured at a wavelength of 450 nm using a VersaMax® microplate reader.
Cell morphological analysis
H9c2 cells were seeded at a density of 1 × 105 cells/well in 6-well plates, as previously described. 17 When confluence reached 70–80%, the cells were treated with bupivacaine (10-3 M), a lipid emulsion (0.1, 0.2, and 0.3%), and various inhibitors (oxfenicine [2 × 10-3 M], LY-294002 [3 × 10-6 M], and SB216763 [5 × 10-6 M]), alone or in combination. H9c2 cells were treated with bupiavcaine alone for 36 h, lipid emulsion for 1 h followed by bupivcaine for 36 h, lipid emulsion alone for 37 h, the inhibitor for 1 h followed by lipid emulsion for 1 h and post-treatment with bupivacaine for 36 h, or inhibitor alone for 38 h. After washing twice with phosphate buffered saline (PBS), the morphology of H9c2 cells was photographed under a phase-contrast microscope (Olympus Optical Co. Ltd, Fluoview 500, Tokyo, Japan).
Measurement of intracellular levels of reactive oxygen species
The generation of intracellular ROS was measured using dichlorodihydrofluorescein (H2DCFDA, Calbiochem, San Diego, CA, USA), as previously described. 18 H9c2 cells (5 × 103 cells/100 μL) grown on glass-bottom culture dishes (SPL, Pocheon, Korea) were treated with 10-3 M bupivacaine alone for 24 h, 0.1% lipid emulsion or inhibitor (10-3 M NAC, and 3 × 10-6 M mitotempo) for 1 h followed by 10-3 M bupivacaine for 24 h. H9c2 cells were then incubated with 5 μM H2DCFDA at 37°C for 30 min in the dark. After incubation, the cells were washed thrice with 1× PBS. Cell fluorescence was immediately analyzed using an IX70 Fluoview (Olympus, Tokyo, Japan). For the detection of green fluorescence (H2DCFDA), cells were illuminated with 488 and 518 nm laser lines. Fluorescent images were analyzed using the Fluoview software program (version 2.0, Olympus, Tokyo, Japan).
Measurement of mitochondrial membrane potential
Changes in the mitochondrial membrane potential (MMP) were determined using the JC-1 MMP detection kit (Biotium Inc. Hayward, CA, USA), as previously described. 18 Briefly, H9c2 cells (5 × 103 cells/100 μL), grown on glass-bottom culture dishes (SPL), were treated with 10-3 M bupivacaine alone for 24 h, or 0.1% lipid emulsion or inhibitor (10-3 M NAC, and 3 × 10-6 M mitotempo) for 1 h followed by 10-3 M bupivacaine for 24 h. Then H9c2 cells were stained with 1× JC-1 reagent at 37°C for 15 min, and resuspended in 1× PBS. Changes in the MMP were measured at the single cell level using fluorescence image analysis. Mitochondrial function was usually monitored with changes in the fluorescence intensity ratio (red/green) using a GloMax explorer (Promega, Madison, WI, USA).
Measurement of intracellular calcium concentration
The concentration of intracellular calcium [Ca2+]i was measured using a confocal laser scanning microscope equipped with a fluorescence system (IX70 Fluoview, Olympus), as previously described. 18 H9c2 cells cultured on glass-bottom culture dishes (SPL) were incubated with 5 μM Fluo-3AM in serum-free DMEM media for 30 min and washed thrice with 1× PBS. Each fluorescent image was scanned every 5 s at excitation and emission wavelengths of 488 and 530 nm, respectively. All scanned images were processed to analyze the changes in [Ca2+]i at the single-cell level. In each cell studied, the changes in [Ca2+]i were calculated as fluorescence intensity (F) divided by the basal fluorescence intensity before treatment (F0) to control for variations in basal fluorescence (F/F0). Net changes in F were represented as (Fmax − F0)/F0, where Fmax is the maximum level of fluorescence intensity after the addition of chemicals. As changes in [Ca2+]i are an immediate reaction in response to chemicals (10-3 M bupivacaine, 0.1% lipid emulsion, 10-3 M NAC, and 3 × 10-6 M mitotempo), they were measured for 8 min after treatment with chemicals.
Materials
Chemicals were of the highest purity and commercially available. In particular, 20% Intralipid® was obtained from Fresenius Kabi AB (Uppsala, Sweden). Bupivacaine and levobupivacaine were obtained from Myungmoon Pharmaceutical Company (Seoul, Republic of Korea) and AbbVie Inc (NorthChicago, Illinois, USA), respectively. Ropivacaine and mepivacaine were obtained from Mitsubishi Tanabe Pharmaceutical Korea Company (Gyeonggi-do, Republic of Korea) and Reyon Pharmaceutical Company (Seoul, Republic of Korea), respectively. SB216763 was obtained from Tocris Bioscience (Bristol, UK). NAC, mitotempo, and LY-294,002 were obtained from Sigma Aldrich (St Louis, MO, USA). Oxfenicine was obtained from Tokyo Chemical Industry (Tokyo, Japan). SB216763 and LY-294002 were dissolved in DMSO (final concentration of DMSO: 0.11%). Other drugs were dissolved in distilled water.
Statistical analysis
Data are presented as mean ± standard deviation (SD). The effect of local anesthetics and the lipid emulsion, alone or combined, on the activities of CPT-I, CACT, and CPT-II and cell viability were analyzed using one-way analysis of variance (ANOVA) followed by Bonferroni’s test or Kruskal–Wallis test followed by Dunn’s multiple comparison test (Prism, version 5.0; GraphPad Software, San Diego, CA, USA). The effect of lipid emulsion on the concentration of each local anesthetic in Krebs solution was analyzed using the Kruskal–Wallis test followed by Dunn’s multiple comparison test. The effect of lipid emulsion on the activity of CACT was estimated using the standardized mean difference (SMD). Briefly, SMD was calculated using the following equation: the mean difference in the activity of CACT between two groups divided by the pooled standard deviation. 19 The correlation between log P (log[octanol/water partition efficient]) and lipid emulsion-mediated reduction in the concentrations of local anesthetics was analyzed using the Spearman correlation coefficient. The Shapiro-Wilk test was used to evaluate the normality of data regarding ROS, the MMP, and intracellular calcium concentration. The effects of bupivacaine, lipid emulsions, and inhibitors on ROS, the MMP, and intracellular calcium concentration were analyzed using one-way ANOVA followed by Bonferroni’s test or Kruskal–Wallis test followed by Dunn’s multiple comparison test (OriginPro2020, OriginLab Corp., Northampton, MA, USA). p < 0.05 was considered statistically significant.
Results
The activity of CPT-I and CPT-II in H9c2 cells was not significantly altered by toxic concentrations of levobupivacaine, ropivacaine and mepivacaine (Figure 1 and 2). However, the activity of CACT in H9c2 cells was reduced by toxic concentrations of levobupivacaine, bupivacaine, ropivacaine, and mepivacaine (Figure 3; p < 0.05 versus control). The SMD values of levobupivacaine, bupivacaine, ropivacaine, and mepivacaine was found to be - 10.09 (95% confidence interval [CI]: −14.29 to −5.90), −8.07 (95% CI: −13.99 to −2.14), −3.54 (95% CI: −5.35 to −1.73), and −8.28 (95% CI: −13.22 to −3.23), respectively. The levobupivacaine- and bupivacaine-induced inhibition of CACT was reversed by all concentrations of lipid emulsion (Figure 3; p < 0.001 versus local anesthetic alone; SMD of levobupivcaine: 0.05%; 3.00 [95% CI: 1.35 to 4.65], 0.1%; 7.59 [95% CI: 4.35 to 10.83], and 0.2%; 7.92 [95% CI: 4.56 to 11.29]; SMD of bupivcaine: 0.05%; 8.03 [95% CI: 3.86 to 12.20], 0.1%; 22.13 [95% CI: 11.20 to 33.06], and 0.2%; 21.18 [95% CI: 10.71 to 36.65]). The ropivacaine-induced inhibition of CACT was reversed by moderate and high concentrations of lipid emulsion (Figure 3; p < 0.01 versus ropivacaine alone; SMD: 0.1%; 1.88 [95% CI: 0.52 to 3.23], and 0.2%; 3.52 [95% CI: 1.71 to 5.32]). In contrast, the mepivacaine-induced inhibition of CACT was not significantly altered by the highest concentration of lipid emulsion (Figure 3). In addition, the activity of CACT in H9c2 cells was greatly increased by all concentrations of lipid emulsion alone (Figure 3: p < 0.05 versus control; SMD at 0.05, 0.1, and 0.2% lipid emulsion: 4.05 [95% CI: 2.07 to 6.03], 2.22 [95% CI: 0.78 to 3.66], and 1.78 [95% CI: 0.45 to 3.12], respectively). Effects of toxic doses of local anesthetics on carnitine palmitoyltransferase-I (CPT-I) activity in H9c2 rat cardiomyoblasts. Control: without local anesthetic. Data (N = 5) are shown as mean ± SD. N indicates the number of independent experiments. Effects of toxic doses of local anesthetics on carnitine palmitoyltransferase-II (CPT- II) activity in H9c2 rat cardiomyoblasts. Control: without local anesthetic. Data (N = 4) are presented as mean ± SD. N indicates the number of independent experiments. Effects of the lipid emulsion (LE) and toxic doses of local anesthetic (LA), alone or combined, on carnitine acylcarnitine translocase (CACT) activity in H9c2 rat cardiomyoblasts. Control: without LA or LE. Data are shown as mean ± SD. *p < 0.05, **p < 0.01, and ***p < 0.001 versus control. ††p < 0.01, and †††p < 0.001 versus LA alone. N (levobupivacaine [N=6], bupivacaine [N=4], mepivacaine [N=3], and LE alone [N=6]) indicates the number of experiments.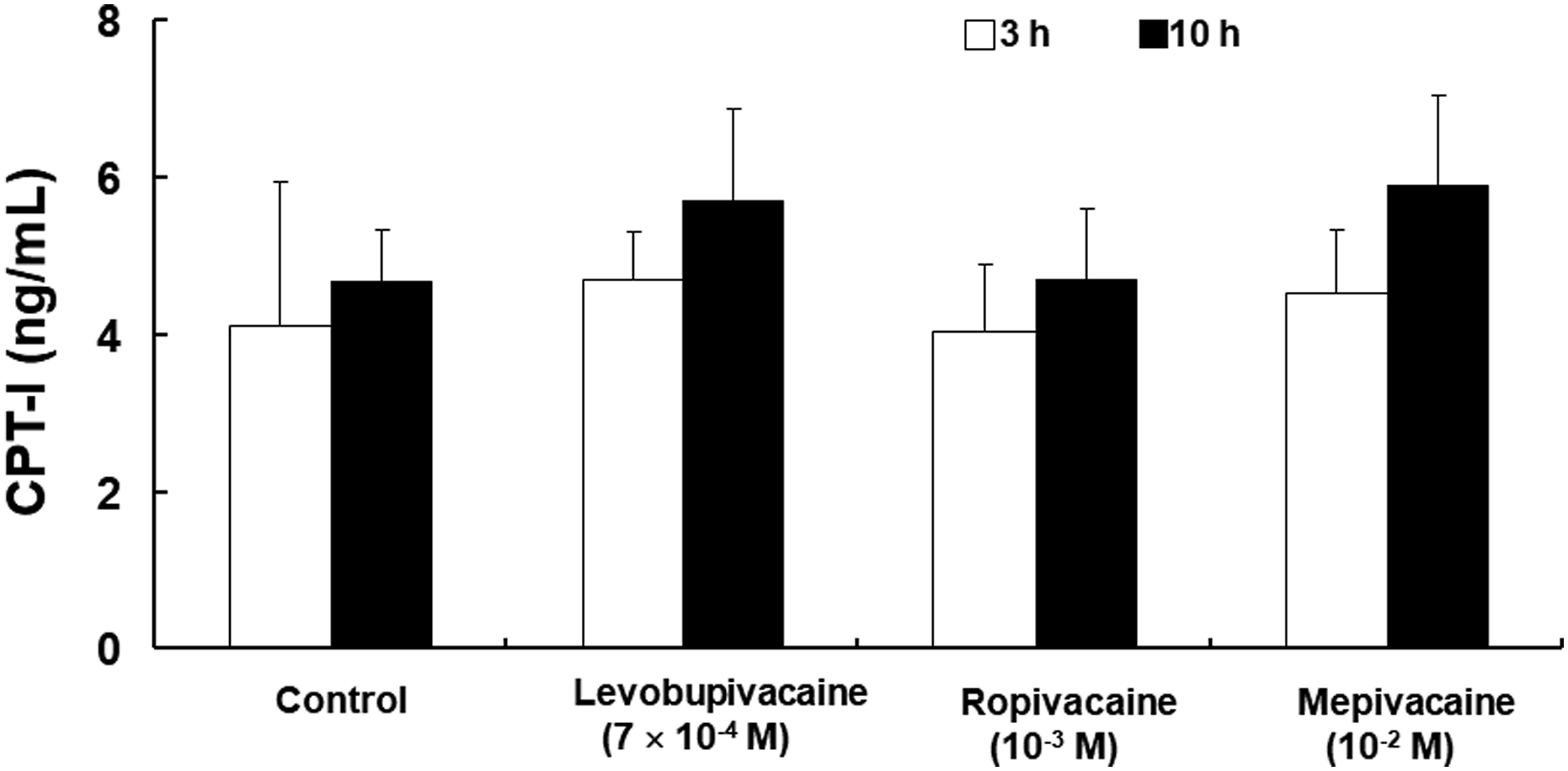


The concentration of levobupivacaine in Krebs solution was slightly decreased by the highest concentration of lipid emulsion (Figure 4(a)); p < 0.05 versus levobupivacaine alone). Contrastingly, the concentration of ropivacaine was not significantly altered by lipid emulsion (Figure 4(b)). In addition, the concentration of mepivacaine in Krebs solution was not affected by lipid emulsion (Figure 4(c)). The Spearman correlation coefficient between log P (levobupivacaine = 3.2, ropivacaine = 2.9, and mepivacaine = 1.95) and lipid emulsion (2%)-mediated reduction (%) in the concentration of local anesthetics was found to be 0.652 (p < 0.05). Effects of the lipid emulsion (LE) on the toxic concentrations of levobupivacaine (LBV, a), ropivacaine (RPV, b), and mepivacaine (MPV, c) in Krebs solution. Data are shown as mean ± SD and expressed as the percentage of the concentration of each local anesthetic alone without LE (control). Control: local anesthetic alone. *p < 0.05 versus control (local anesthetic alone). a: Experiment was repeated five, five, four, and four times for the control and 0.05, 0.1, and 0.2% of the LE, respectively. b: Experiment was repeated five, five, five, and four times for the control and 0.05, 0.1, and 0.2% of the LE, respectively. c: Experiment was repeated five times.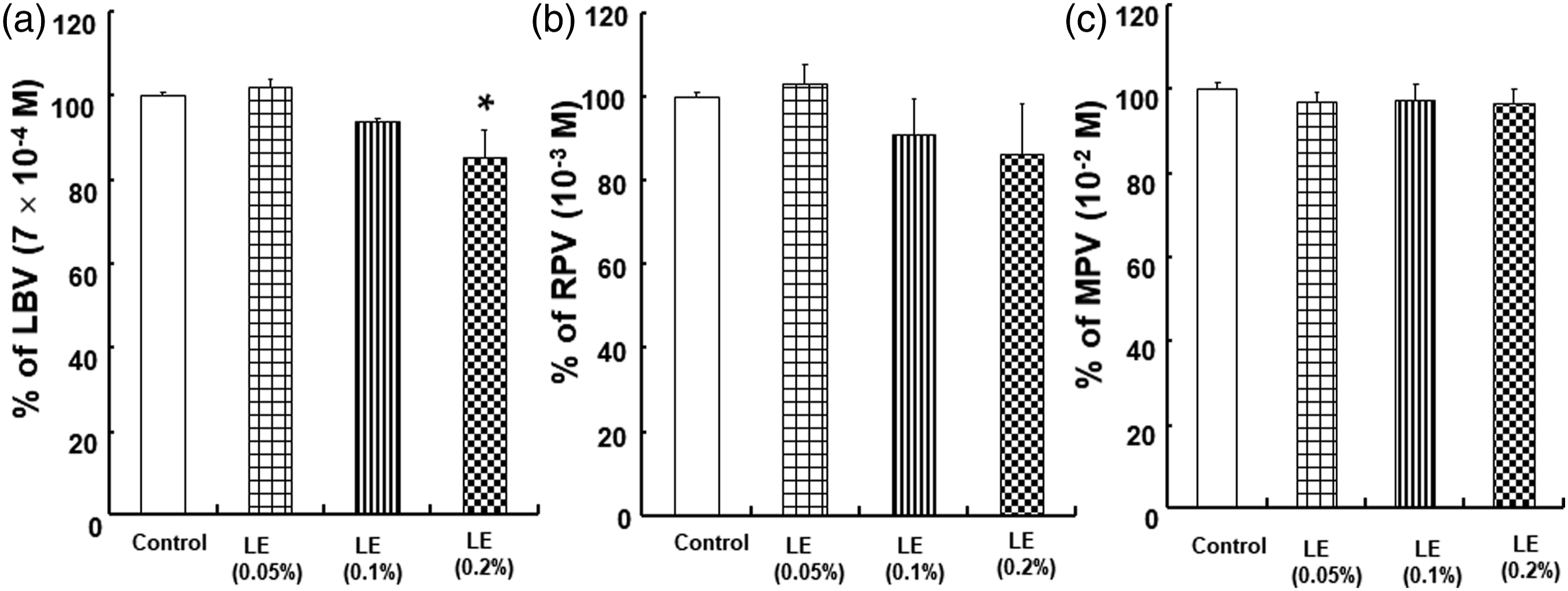
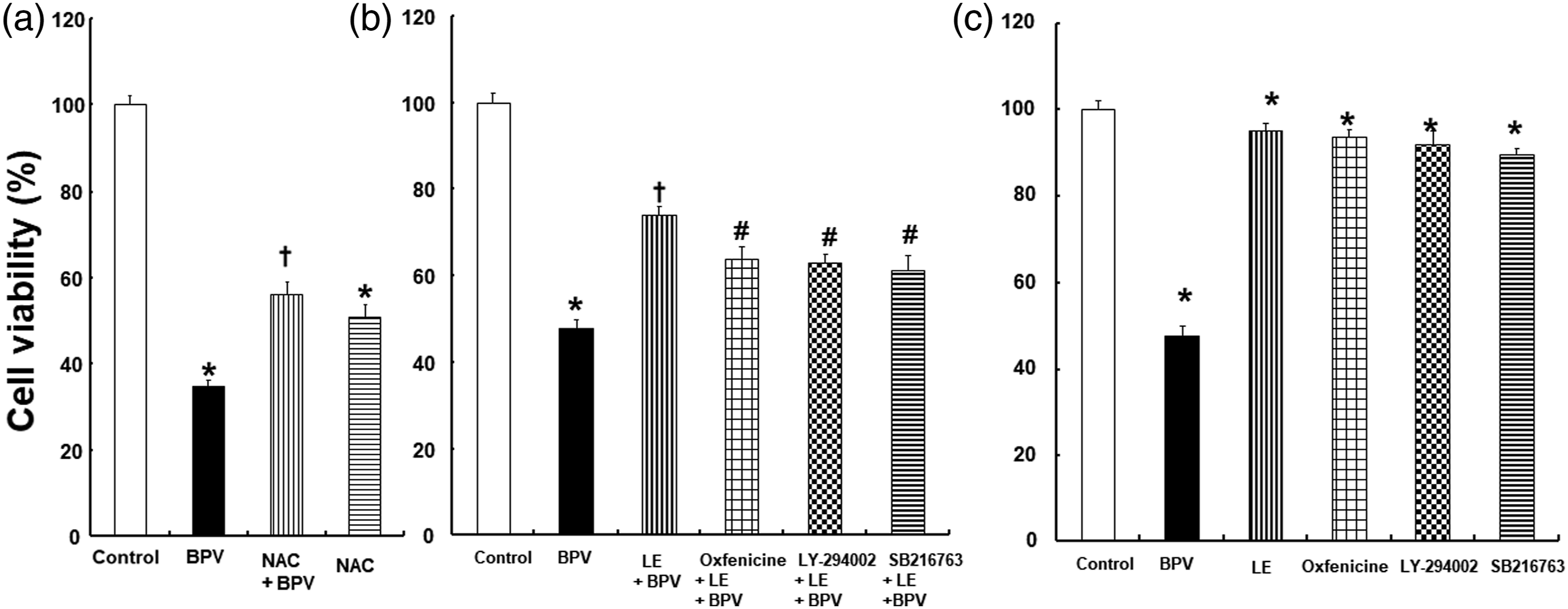
The viability of H9c2 cells was decreased by bupivacaine (Figure 5(a)); p < 0.001 versus control), whereas the bupivacaine-induced decrease in the viability of H9c2 cells was attenuated by lipid emulsion (Figure 5(a)); p < 0.001 versus bupivacaine alone). The toxic dose of bupivacaine-induced decreased cell viability was attenuated by NAC (Figure 6(a)); p < 0.001 versus bupivacaine alone). In addition, the lipid emulsion-mediated inhibition of bupivacaine-induced decreased cell viability was attenuated by the CPT-I inhibitor oxfenicine, phosphoinositide 3-kinase inhibitor LY-294002, and glycogen synthase kinase-3β inhibitor SB216763 (Figure 6(b)); p < 0.001 versus lipid emulsion + bupivacaine). Cell density was reduced by bupivacaine (Figure 7(a)). However, bupivacaine-induced cellular morphological changes were diminished by lipid emulsion (Figure 7(a))). In turn, the lipid emulsion-induced inhibition of the bupivacaine-induced morphological changes was attenuated by oxfenicine, LY-294002, and SB216763 (Figure 7(b)). However, DMSO, which was used for the dissolution of LY-294002 and SB216763, had no effect on cell viability (Supplementary Figure 1). Effects of the toxic dose of bupivacaine (BPV, 10-3 M) and lipid emulsion (LE), combined (a) or alone (b), on the viability of H9c2 rat cardiomyoblasts. Control: without pretreatment. Data (N = 4) are shown as mean ± SD. N indicates the number of independent experiments. *p < 0.001 versus control. †p < 0.001 versus BPV alone. a: Effects of bupivacaine (BPV, 2 × 10-3 M) and N-acetyl-L-cysteine (NAC, 3 × 10-2 M), alone or combined, on the viability of H9c2 rat cardiomyoblasts. Control: without pretreatment. Data (N = 3) are shown as mean ± SD. N indicates the number of independent experiments. *p < 0.001 versus control. †p < 0.001 versus BPV alone. b and c: Effects of BPV (10-3 M), the lipid emulsion (LE, 0.1%), inhibitors (oxfenicine [2 × 10-3 M], LY-294002 [3 × 10-6 M], and SB216763 [5 × 10-6 M]), alone or combined, on the viability of H9c2 rat cardiomyoblasts. Control: without pretreatment. Data (N = 3) are shown as mean ± SD. N indicates the number of independent experiments. *p < 0.001 versus control. †p < 0.001 versus BPV alone. #p < 0.001 versus LE + BPV. a: Representative phase-contrast microscopic images of H9c2 rat cardiomyoblasts after treatment with bupivacaine (BPV, 10-3 M) and lipid emulsion (LE), alone or in combination. Control: without pretreatment. b: Representative phase-contrast microscopic images of H9c2 rat cardiomyoblasts after treatment with BPV (10-3 M), LE, and various inhibitors (oxfenicine [2 × 10-3 M], LY-294002 [3 × 10-6 M], and SB216763 [5 × 10-6 M]), alone or combined. Control: without pretreatment.

The production of ROS in H9c2 cells was increased by bupivacaine (Figure 8; p < 0.001 versus control), whereas the bupivacaine-induced increase in the production of ROS was inhibited by the lipid emulsion, the ROS scavenger NAC, and the mitochondrial ROS scavenger mitotempo (Figure 8; p < 0.001 versus bupivacaine alone). The MMP of H9c2 cells was decreased by bupivacaine (Figure 9; p < 0.001 versus control). However, the bupivacaine-induced decrease in the MMP was considerably attenuated by lipid emulsion, NAC, and mitotempo (Figure 9; p < 0.001 versus bupivacaine alone). In addition, the intracellular concentration of calcium in H9c2 cells was increased by bupivacaine (Figure 10; p < 0.001 versus control), whereas this bupivacaine -induced increase in intracellular calcium concentration in H9c2 cells was reduced by the lipid emulsion, NAC, and mitotempo (Figure 10; p < 0.05 versus bupivacaine alone). Effects of the lipid emulsion (LE, 0.1%), N-acetyl-L-cysteine (NAC, 10-3 M), and mitotempo (3 × 10-6 M) on the bupivacaine (BPV, 10-3 M)-induced production of reactive oxygen species (ROS) in H9c2 rat cardiomyoblasts. The intracellular levels of ROS were quantified by measuring the fluorescence intensity (FI) using fluorescence microscopy. Control: without pretreatment. Each bar represents the mean ± SD of three independent experiments. *p < 0.001 versus control. †p < 0.001 versus BPV alone. Effects of the lipid emulsion (LE, 0.1%), N-acetyl-L-cysteine (NAC, 10-3 M), and mitotempo (3 × 10-6 M) on the bupivacaine (BPV, 10-3 M)-induced changes in the mitochondrial membrane potential (MMP) of H9c2 rat cardiomyoblasts. Green and red indicate JC-1 monomers and JC-1 aggregates, respectively. The scale bar represents 50 μm. Control: without pretreatment. Each bar represents the mean ± SD of three independent experiments. FI: fluorescence intensity. *p < 0.01 versus control. †p < 0.001 versus BPV alone. Effects of the lipid emulsion (LE, 0.1%), N-acetyl-L-cysteine (NAC, 10-3 M), and mitotempo (3 × 10-6 M) on the bupivacaine (BPV, 10-3 M)-induced changes in the concentration of intracellular calcium ([Ca2+]i) in H9c2 rat cardiomyoblasts. Control: without pretreatment. Each bar represents the mean ± SD of three independent experiments. *p < 0.05, †p < 0.01 and #p< 0.001 versus BPV alone.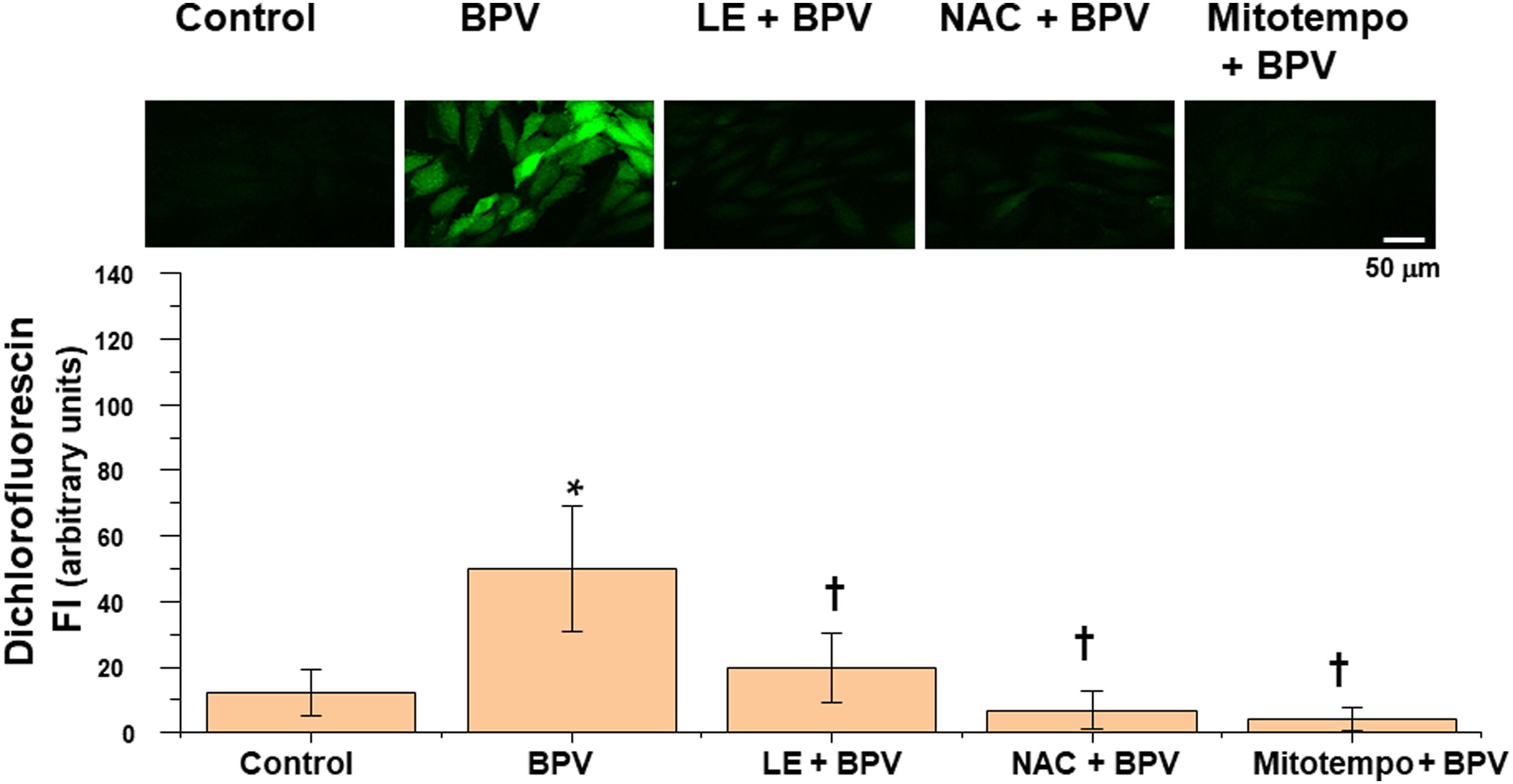


Discussion
This is the first study to suggest that a lipid emulsion could attenuate the levobupivacaine-induced inhibition of CACT, potentially owing to the sequestration of levobupivacaine. In addition, the same lipid emulsion inhibited the bupivacaine-induced mitochondrial dysfunctions by attenuating the production of ROS.
Three steps are involved in the transportation of long-chain fatty acids, which are the preferential energy source used in the heart, from the cytoplasm to the mitochondrial matrix in heart cells. 6 First, long chain fatty acids are transported to the cytoplasm via fatty acid transport proteins of the plasma membrane. There, long chain fatty acyl-CoA synthase catalyzes the production of long chain fatty acyl-CoA from long chain fatty acids. 6 Subsequently, CPT-I catalyzes the production of long chain fatty acylcarnitine from long chain fatty acyl-CoA and carnitine, which is transported via the organic carnitine transporter novel type 2 of plasma membrane. 6 In turn, long chain fatty acylcarnitine is transported from the cytoplasm to the inner side of the outer mitochondrial membrane. 6 Second, long chain fatty acylcarnitine is transported from the intermembrane space of mitochondria to the mitochondrial matrix by CACT. 6 Third, long chain fatty acylcarnitine is split to carnitine and long chain fatty acyl-CoA by CPT-II. 6 Then, carnitine is returned to the cytoplasm by CACT, and long chain fatty acyl-CoA undergoes beta-oxidation in the mitochondrial matrix to produce ATP. 6 Intralipid (20%) containing only 100% long-chain fatty acids requires a carnitine shuttle for its transportation from the cytoplasm to the mitochondrial matrix.20,21 Pretreatment with L-carnitine, which is known to be associated with carnitine shuttles, has been reported to decrease the susceptibility to bupivacaine-induced cardiotoxicity. 22 In addition, L-carnitine deficiency was shown to decrease the time to bupivacaine-induced cardiac arrest, whereas L-carnitine repletion prolonged the time to bupivacaine-induced cardiac arrest in L-carnitine deficiency group. 23 Similar to the results of previous report, the toxic doses of levobupivacaine (7 × 10-4 M), ropivacaine (10-3 M), and mepivacaine (10-2 M) inhibited the activity of CACT. 7 Oxfenicine did not change myocardial contractility, but a toxic dose of bupivacaine (5 × 10-4 M) in the presence of oxfenicine produced asystole, suggesting that bupivacaine-induced asystole seems to not be mediated by CPT-I. 24 Taken together, as the toxic doses of levobupivacaine, ropivacaine, and mepivacaine did not significantly alter the activities of CPT-I and CPT-II, cardiotoxicity induced by the toxic doses of aminoamide local anesthetics seems to be mediated partially by the inhibition of CACT in cardiac mitochondria. All concentrations of the lipid emulsion attenuated the levobupivacaine- and bupivacaine-induced inhibition of CACT (Figure 3). On the contrary, only high and medium concentrations of the lipid emulsion (0.1 and 0.2%) attenuated the ropivacaine (10-3 M)-mediated inhibition of CACT (Figure 3). However, the highest concentration of the lipid emulsion did not significantly affect the mepivacaine (10-2 M)-induced inhibition of CACT (Figure 3). These results suggested that the potency of the lipid emulsion-mediated attenuation of the local anesthetic-induced inhibition of CACT seems to be dependent on the lipid solubility of local anesthetics. 25 It has also been reported that the lipid emulsion has higher binding affinity to levobupivacaine than to ropivacaine. 26 In addition, the magnitude of the lipid emulsion-extracted local anesthetics was found to be: bupivacaine > ropivacaine > mepivacaine. 27 In agreement with previous reports, the highest concentration of the lipid emulsion (0.2%), which corresponds to one fifth of its plasma concentration (final concentration of plasma triglycerides: 1%) used to treat cardiac arrest induced by a toxic dose of bupivacaine in an in vivo experiment, was found to slightly decrease the concentration of levobupivacaine (7 × 10-4 M) (Figure 4(a)), whereas it did not significantly alter the concentrations of ropivacaine (10-4 M) and mepivacaine (10-2 M) (Figure 4(b) and (c)).3,26–28 Taking into consideration the above-mentioned results, the lipid emulsion-mediated attenuation of the local anesthetics-induced inhibition of CACT seems to be partially associated with the absorption of local anesthetics into the lipid phase of the emulsion in a lipid solubility-dependent manner (Spearman correlation coefficient: 0.652). In addition, the lipid emulsion (0.05–0.2%) (effect size: 4.05 to 1.78) alone profoundly increased the activity of CACT, potentially owing to the enhanced supply of long-chain fatty acids from intralipid (Figure 3). Thus, further studies are needed to determine whether the lipid emulsion-mediated amelioration of CACT inhibition induced by toxic doses of local anesthetics is associated with either the restoration (indirect effect) of the inhibited activity of CACT due to a reduction in the concentration of local anesthetics or the abundant supply (direct effect) of long-chain fatty acylcarnitine (produced by Intralipid), a substrate used by CACT.
As lipid emulsion-mediated amelioration of CACT inhibition was higher in bupivacaine than in levobupivacaine (Figure 3), we examined the effects of lipid emulsion on mitochondrial dysfunction and decreased cell viability induced by toxic concentrations of bupivacaine. Furthermore, the lipid emulsion was found to attenuate the bupivacaine-induced decreased cell viability (Figure 6(b)), which was consistent with the observed morphological changes, such as the lipid emulsion-mediated attenuation of the bupivacaine-induced decreased cell density (Figure 7(a)). In a previous study, the lipid emulsion was found to attenuate the bupivacaine-induced apoptosis of cells via the reduction of the intracellular oxidative stress. 12 Bupivacaine was reported to depress both complexes I and III of the mitochondrial respiratory chain and lead to increased ROS. 29 In agreement with these reports, the lipid emulsion in our study inhibited the bupivacaine (10-3 M)-induced production of ROS (Figure 8).12,29 Conclusively, as the ROS scavenger NAC and mitochondrial ROS scavenger mitotempo inhibited the bupivacaine (10-3 M)-induced production of ROS (Figure 8) and NAC inhibited the bupivacaine-induced decrease in cell viability (Figure 6(a))), the lipid emulsion-mediated attenuation of the bupivacaine-induced increase in ROS (Figure 8) seems to contribute to the lipid emulsion-mediated attenuation of the bupivacaine-induced decrease in cell viability (Figure 6(b)).12,29 As NAC and mitotempo inhibited the bupivacaine-induced depolarization of mitochondrial membrane potential (Figure 9), this depolarization appears to have been caused by the mitochondrial production of ROS. Similarly, the bupivacaine-induced increase in intracellular calcium (Figure 10) seems to have been associated with the production of ROS. The CPT-I inhibitor oxfenicine slightly attenuated the lipid emulsion-mediated recovery of the bupivacaine (10-3 M)-induced decrease in cell viability (Figure 6(b)), suggesting that this lipid emulsion-mediated cell viability recovery is associated with the restoration of the activity of CPT-I. The lipid emulsion has also been reported to attenuate ischemia and reperfusion injury via the inhibition of the opening of mitochondrial permeability transition pores, which is mediated through phosphoinositide-3 kinase and glycogen synthase kinase-3β induced by protein kinase B and extracellular signal-regulated kinase. 30 Consistently, the phosphoinositide-3 kinase inhibitor LY-294,002 and glycogen synthase kinase-3β inhibitor SB216763 slightly attenuated the lipid emulsion-mediated inhibition of the bupivacaine (10-3 M)-induced decrease in cell viability (Figure 6(b)). 30 This slightly protective effect of lipid emulsion might be due to the activation of pathways involving the phosphorylation of phosphoinositide-3 kinase and glycogen synthase kinase-3β.
Carnitine deficiency was reported to produce non-toxic dose bupivacaine-induced ventricular arrhythmia, potentially due to the relatively increased susceptibility to bupivacaine toxicity through a relatively enhanced inhibition of CACT by non-toxic doses of bupivacaine.7,31 In addition, the lipid emulsion was reported to treat non-toxic dose bupivacaine-induced hypotension in patients with secondary carnitine deficiency. 32 Extrapolating these result into the clinical practice would suggest that treatment with lipid emulsion might be effective in alleviating the cardiovascular collapse induced by bupivacaine or levobupivacaine in patients with carnitine deficiency, because the lipid emulsion reversed the levobupivacaine- or bupivacaine-induced decrease in the activity of CACT and increase in the production of ROS. There are some limitations to this study. First, we used rat cardiomyoblasts instead of human cardiomyocytes. Second, this study was an in vitro study, whereas an in vivo study involving the determination of the activities of CPT-I, CACT, and CPT-II as well as the levels of ROS in the heart and those of local anesthetics in the plasma would be more clinically relevant. Third, we used a lipid emulsion pretreatment, whereas lipid emulsion post-treatment would be clinically used against local anesthetic toxicity. Fourth, ELISA was performed to determine CACT activity using culture media (supernatants) of H9c2 cells treated with local anesthetic and lipid emulsion, whereas high performance liquid chromatography was performed to determine local anesthetic concentration using a Krebs solution containing local anesthetic and lipid emulsion.
In conclusion, our results suggested that the lipid emulsion attenuated the local anesthetic-mediated inhibition of CACT in a lipid solubility-dependent manner (levobupivacaine > ropivacaine > mepivacaine), probably owing to the lipid emulsion-mediated reduction in the concentration of local anesthetics. In addition, the lipid emulsion attenuated the bupivacaine-induced mitochondrial dysfunctions via the inhibition of ROS production.
Supplemental Material
sj-pdf-1-het-10.1177_09603271211065978 – Supplemental Material for Lipid emulsions attenuate the inhibition of carnitine acylcarnitine translocase induced by toxic doses of local anesthetics in rat cardiomyoblasts
Supplemental Material, sj-pdf-1-het-10.1177_09603271211065978 for Lipid emulsions attenuate the inhibition of carnitine acylcarnitine translocase induced by toxic doses of local anesthetics in rat cardiomyoblasts by Seong-Ho Ok, Dawon Kang, Soo H Lee, Hyun-Jin Kim, Seung H Ahn, and Ju-Tae Sohn in Human & Experimental Toxicology
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Research Foundation of Korea(NRF) grant funded by the Korea government (MSIT) (NRF-2021R1F1A1062363) and biomedical research institute fund (GNUHBRIF-2021-0010) from the Gyeongsang National University Hospital.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.