
Abstract
Local anesthetic toxicity is thought to be mediated partly by inhibition of cardiac mitochondrial function. Intravenous (i.v.) lipid emulsion may overcome this energy depletion, but doses larger than currently recommended may be needed for rescue effect. In this randomized study with anesthetized pigs, we compared the effect of a large dose, 4 mL/kg, of i.v. 20% Intralipid® (n = 7) with Ringer’s acetate (n = 6) on cardiovascular recovery after a cardiotoxic dose of bupivacaine. We also examined mitochondrial respiratory function in myocardial cell homogenates analyzed promptly after needle biopsies from the animals. Bupivacaine plasma concentrations were quantified from plasma samples. Arterial blood pressure recovered faster and systemic vascular resistance rose more rapidly after Intralipid than Ringer’s acetate administration (p < 0.0001), but Intralipid did not increase cardiac index or left ventricular ejection fraction. The lipid-based mitochondrial respiration was stimulated by approximately 30% after Intralipid (p < 0.05) but unaffected by Ringer’s acetate. The mean (standard deviation) area under the concentration–time curve (AUC) of total bupivacaine was greater after Intralipid (105.2 (13.6) mg·min/L) than after Ringer’s acetate (88.1 (7.1) mg·min/L) (p = 0.019). After Intralipid, the AUC of the lipid-un-entrapped bupivacaine portion (97.0 (14.5) mg·min/L) was 8% lower than that of total bupivacaine (p < 0.0001). To conclude, 4 mL/kg of Intralipid expedited cardiovascular recovery from bupivacaine cardiotoxicity mainly by increasing systemic vascular resistance. The increased myocardial mitochondrial respiration and bupivacaine entrapment after Intralipid did not improve cardiac function.
Introduction
Local anesthetic systemic toxicity is a rare 1 but potentially life-threatening complication of regional anesthesia. 2 Within the last decade, intravenous (i.v.) lipid emulsion has been adopted as a recommended treatment of local anesthetic toxicity 3 even though its impact on therapeutic outcome is not evidence based and its mechanisms of action are largely unknown. 4 The initially hypothesized mechanism was the formation of a “lipid sink” that entraps lipophilic molecules into an expanded plasma lipid phase, reversing toxicity. 5,6 Later, other mechanisms such as an effect on tissue distribution, 7 –9 myocardial ion channels, 10 or cardiotony 11 have been suggested. Lipid emulsion may also protect myocardial cells from local anesthetic-induced apoptosis. 12 Another proposed mechanism is a metabolic benefit of lipid emulsion that reverses local anesthetic-induced inhibition of mitochondrial respiration in cardiac tissue. 13 In isolated myocardial mitochondria, the main depressive effect of bupivacaine on the mitochondrial respiratory chain is the inhibition of complex I (nicotinamide adenine dinucleotide (NADH) dehydrogenase). 14,15 Supporting this metabolic theory, the administration of fatty acid β-oxidation inhibitor prevents the resuscitative effect of lipid emulsion after bupivacaine toxicity in rats. 16
The objective of this investigation was to determine the effect of a larger dose of lipid emulsion than recommended on cardiac lipid oxidation and mitochondrial respiration in the heart after bupivacaine toxicity with a hypothesis that lipid emulsion would improve the mitochondrial respiration. We also studied whether the effect of lipid emulsion on energy production correlates with the cardiac function and hemodynamic performance, and whether lipid emulsion affects bupivacaine pharmacokinetics. We deliberately chose to use a larger dose of lipid emulsion than that recommended in guidelines of regional anesthesia societies, 3 since in animal experiments on local anesthetic toxicity, larger lipid doses have been required for a rescue effect. 11,17
Materials and methods
The study protocol was approved by the National Animal Experiment Board (ESAVI/5027/04.10.07/2014; Hämeenlinna, Finland). All experiments were performed in the Research and Development Unit of the Helsinki University Hospital, Helsinki, Finland.
Preparations
Thirteen landrace pigs weighing 23–30 kg (mean 27.5 kg) were used. The estimation of body surface area was based on the weight. 18 Anesthesia was induced using 5% isoflurane. After tracheal intubation, the pigs were mechanically ventilated with 2% isoflurane in 21% oxygen. The respiratory rate was fixed at 20 breaths/min, and the ventilation volume was adjusted to maintain end-tidal CO2 between 5.0% and 5.5%. Esophageal temperature was maintained between 37.5°C and 39.0°C with an external radiant heater and warming mattresses. A two-lumen central venous catheter (7 Fr.; Arrow International, Inc., Reading, Pennsylvania, USA) was inserted into one of the internal jugular veins. An arterial cannula for blood sampling was inserted into the femoral artery, and a PiCCO® Catheter (5F; Pulsion Medical Systems SE, Feldkirchen, Germany) for cardiac output and other hemodynamic measurements was inserted into the other femoral artery. After cannulations, end-expiratory pressure was set to 5 cm H2O, and a lateral thoracotomy was performed to expose the wall of the left ventricle of the heart.
Measurements
Central venous pressures, peripheral oxygen saturation from the tail, and five-lead electrocardiogram (ECG) were continuously monitored with a multimodular patient monitor (Datex-Ohmeda Division; Instrumentarium Co., Helsinki, Finland) and digitally stored using data collection software (iCentral® and S/5 Collect®; GE Healthcare, Helsinki, Finland). Systemic arterial pressure, continuous cardiac output, and systemic vascular resistance were monitored and recorded using a PiCCO PulsionFlex® monitor (V4.0.0.7 A; Pulsion Medical Systems SE).
Serial needle biopsies (mean weight of myocardial biopsies 3.7 mg) were obtained under visual control, from the wall of the left ventricle of the heart using an 18G biopsy needle (Speedcut®; Gallini S.r.l., Mantova, Italy) to assay mitochondrial respiration at predefined time points (Figure 1). Similarly, serial arterial blood samples, and at the end of experiment, tissue samples from the lungs and the apex of the heart were collected for bupivacaine quantification.

Study protocol. After baseline samples and values were obtained, bupivacaine 2 mg/kg/min was infused i.v. until the MAP decreased to 60% of baseline level. Then, the pigs were randomly given either lipid emulsion or Ringer’s acetate 4 mL/kg in 1 min. Isoflurane administration was discontinued at the end of bupivacaine infusion and continued again at 1% when MAP increased back to 60% and increased to 2% when MAP reached the baseline level. Sampling times are presented in the figure. i.v.: intravenous; MAP: mean arterial pressure.
An experienced cardiac anesthesiologist, blinded to the treatment, recorded epicardial echocardiography from short axis left ventricular view before and during the experiment. The left ventricular ejection fraction (EF) was measured from M-mode acquisition using the Teicholz method. 19
Pilot study—lipid emulsion alone and arterial blood pressure
Five landrace pigs (22–26 kg) were anesthetized with isoflurane in oxygen, intubated, mechanically ventilated, and monitored with continuous 5-lead ECG, peripheral oxygen saturation, central venous pressure (CVP), and femoral arterial blood pressure similarly as in the animals of the main study. After a stabilization period of 30 min and a CVP ≥ 2 mmHg, 4 mL/kg of Ringer’s acetate (Ringer-Acetat Baxter Viaflo®; Baxter Medical, Kista, Sweden) was infused into a jugular vein in 60 s and mean arterial blood pressure was registered. In comparable and stable conditions 60 min later, the same pig received 4 mL/kg of 20% lipid emulsion (Intralipid® 20%; Fresenius Kabi AB, Uppsala, Sweden) i.v. in 60 s and arterial blood pressure was registered.
Experiment protocol
After preparations and baseline measurements, the pigs were given an i.v. infusion of bupivacaine HCl (Bicain®; Orion Pharma, Espoo, Finland) 2 mg/kg/min until the mean arterial pressure (MAP) decreased to 60% of its baseline value. After blood sampling, echocardiography, and biopsy, isoflurane administration was discontinued and the inspired oxygen concentration was increased to 100%. The pigs were then given a 4 mL/kg bolus of either 20% lipid emulsion (Intralipid 20%; n = 7) or Ringer’s acetate solution (n = 6) into a central vein in randomized order.
When MAP recovered to above 60% of baseline level, isoflurane was restarted at a concentration of 1%, and when MAP reached the baseline level, isoflurane was raised back to 2%. If MAP declined below 25 mmHg, open chest cardiac massage was initiated until MAP stayed unsupported over 25 mmHg. At the end of the experiment, tissue samples for bupivacaine concentration quantifications were obtained from the apex of the heart and from the lower lobe of the left lung. Then, the pigs were euthanized with a rapid i.v. bolus of potassium chloride concentrate.
Mitochondrial high-resolution respirometry
After obtaining cardiac biopsies, the samples (mean weight 3.7 mg) were immediately transferred into ice-cold relaxing and biopsy preservating solution BIOPS 20 for transport. Then they were homogenized and the mitochondrial respiration (i.e. adenosine triphosphate (ATP) production) was analyzed using the Oxygraph-2k (OROBOROS Instruments Corp., Innsbruck, Austria) according to the instructions of the manufacturer. 20 The oxygen flux was measured using a previously described fatty acid substrate-uncoupler-inhibitor titration protocol 21 with slight modification in the injection order. The mitochondrial analysis is presented in detail in Online Supplements.
Bupivacaine concentration quantifications
To separate the plasma, the whole-blood samples were centrifuged at 2500g for 10 min and then stored at −22°C. Aliquots of the plasma of the pigs, which received lipid emulsion, were later centrifuged twice at 20,800g for 10 min to separate the lipoid and aqueous fractions. Plasma total bupivacaine concentrations and un-entrapped (nonlipid-bound) bupivacaine concentrations from the further centrifuged lipid-poor plasma were determined using an Agilent 1100 high-performance liquid chromatography (HPLC; Agilent Technologies, Waldbronn, Germany) coupled to an API 2000 HPLC-tandem mass spectrometry system (Sciex Division of MDS Inc., Toronto, Ontario, Canada) with ropivacaine as an internal standard. 22 The quantifications were made during one day, and the coefficients of variation were 10% for plasma samples, 5.4% for heart samples, and 12% for lung samples.
Tissue samples were rinsed by dipping into isotonic saline, dried on filter paper for 5 s and stored at −22°C inside aluminum foil wrap. Later, the samples (0.3 g) were homogenized with an IKA® UltraTurrax T25 homogenizer (Janke & Kunkel, Staufen, Germany) in 4.0 mL of water. The bupivacaine concentrations were determined using the same HPLC method as for the blood samples during one day.
Statistics
After testing for normal distribution, mitochondrial oxygen flux was compared with the unpaired t-test. The changes of each cardiovascular variable after the start of treatment over time were tested separately using repeated measures analysis of variance (ANOVA) with values of the variable over time as within factors, the treatment as the between factor, and their interactions over time. The appropriate F and p values were calculated, and Sidak post hoc testing for multiple comparisons was used for comparisons at different time points. Bupivacaine pharmacokinetics was analyzed by calculating the area under the concentration–time curve from 0 min to 10 min (AUC0–10 min) using the linear trapezoidal rule. The pharmacokinetics was then compared with the paired or unpaired t-test. All values are presented as means and standard deviations (SDs). A p-value < 0.05 was considered significant. All statistical analyses were performed using Prism 6.0 g for Mac OS X (GraphPad Software, Inc., La Jolla, California, USA).
Results
Pilot study
Intravenous lipid emulsion caused an immediate rise in arterial blood pressure (Figure 2) with a return to near baseline in 10 min. The ANOVA analysis of arterial blood pressure revealed an effect of time, and MAP was significantly higher after lipid emulsion than after Ringer’s acetate (p = 0.01) with a significant treatment–time interaction (F (10, 40) = 3.9, p = 0.001).

MAP in pilot study. In the absence of bupivacaine toxicity, administration of 4 mL/kg lipid emulsion i.v. in 1 min increased the arterial blood pressure in pigs. The MAP was significantly higher (p = 0.01) after lipid emulsion than after Ringer’s acetate with also a significant treatment–time interaction (F (10, 40) = 3.9, p = 0.001). In Sidak post-testing, ****p < 0.0001, **p < 0.01, *p < 0.05 between the treatments. i.v.: intravenous; MAP: mean arterial pressure.
Main experiment
The mean (SD) total bupivacaine dose required to decrease MAP to 60% of the baseline level was 8.6 (1.6) mg/kg without differences between groups (p = 0.11). During the scheduled echocardiography and biopsy, immediately after bupivacaine infusion, MAP continued to decrease to a mean of 44% of baseline before either lipid emulsion or Ringer solution infusion were started. One pig in the Ringer group and two pigs in the Lipid group developed pulseless electrical activity soon after the start of rescue solution, necessitating internal cardiac massage throughout the experiment, and were therefore excluded from the hemodynamic analyses. Their MAP was 25 mmHg or less at the time when treatment began. In addition, four pigs receiving Ringer’s solution required internal cardiac massage lasting for 1–4 min, while two pigs receiving lipid emulsion required internal cardiac massage for less than 30 s until MAP remained spontaneously above 25 mmHg. Mild redness of the skin after lipid emulsion administration was observed in three pigs.
Due to technical reasons, the baseline mitochondrial respiration data were lost for one pig in both groups while all mitochondrial respiration data was lost for one pig receiving lipid emulsion resulting in n = 6 in both groups.
Recovery of hemodynamic variables
The ANOVA analysis of arterial blood pressure revealed an effect of time, and although there was no significant difference in MAP between the groups, the treatment–time interaction was significant (F (10, 80) = 7.5, p < 0.0001), consistent with difference in time course of recovery between the treatments. The recovery of MAP back to baseline level took 4 min longer in the Ringer group than in the Lipid group, and post hoc testing revealed a significantly higher MAP in the Lipid group at time points 2–5 min (p < 0.05, Figure 3(a)). Heart rate decreased by approximately 25% in both groups after bupivacaine infusion (data not shown). There was an effect of time, but no difference in the magnitude of heart rate between the groups and no significant treatment–time interaction (F (10, 80) = 1.2, p = 0.32).

MAP and SVRI. After bupivacaine toxicity, lipid emulsion 4 mL/kg in 1 min i.v. improved the recovery of MAP (a); the treatment-time interaction was significant (F (10, 80) = 7.5, p < 0.0001) (b). The increase in arterial blood pressure was accompanied by a significant treatment-time interaction of mean SVRI (F (10, 80) = 6.2, p < 0.0001). In Sidak post-testing, ***p < 0.001, **p < 0.01, *p < 0.05 between the groups. i.v.: intravenous; MAP: mean arterial pressure; SVRI: systemic vascular resistance index.
Similar to arterial blood pressure, the ANOVA analysis of systemic vascular resistance index (SVRI) revealed an effect of time, no difference in SVRI between the groups, but a significant treatment–time interaction (F (10, 80) = 6.2, p < 0.0001; Figure 3(b)): SVRI increased more in pigs receiving lipid emulsion than in pigs receiving Ringer’s solution. Post hoc test revealed significantly higher SVRI in the Lipid group at time points 3–5 min (p < 0.05).
Cardiac index decreased approximately 35% in both groups after bupivacaine infusion and remained reduced throughout the experiment without returning to baseline (Figure 4(a)).The ANOVA analysis of cardiac index revealed an effect of time, no difference in cardiac index between the groups, but a significant treatment–time interaction (F (10, 80) = 2.5, p = 0.01). Cardiac index increased more in the Ringer group, but in post hoc analysis, there was no difference between groups at any time points. EF decreased approximately 50% in both groups after bupivacaine but began to increase in a similar manner in both groups: there was an effect of time, no difference in EF between the groups and no treatment–time interaction (F (3, 30) = 1.2, p = 0.32; Figure 4(b)).
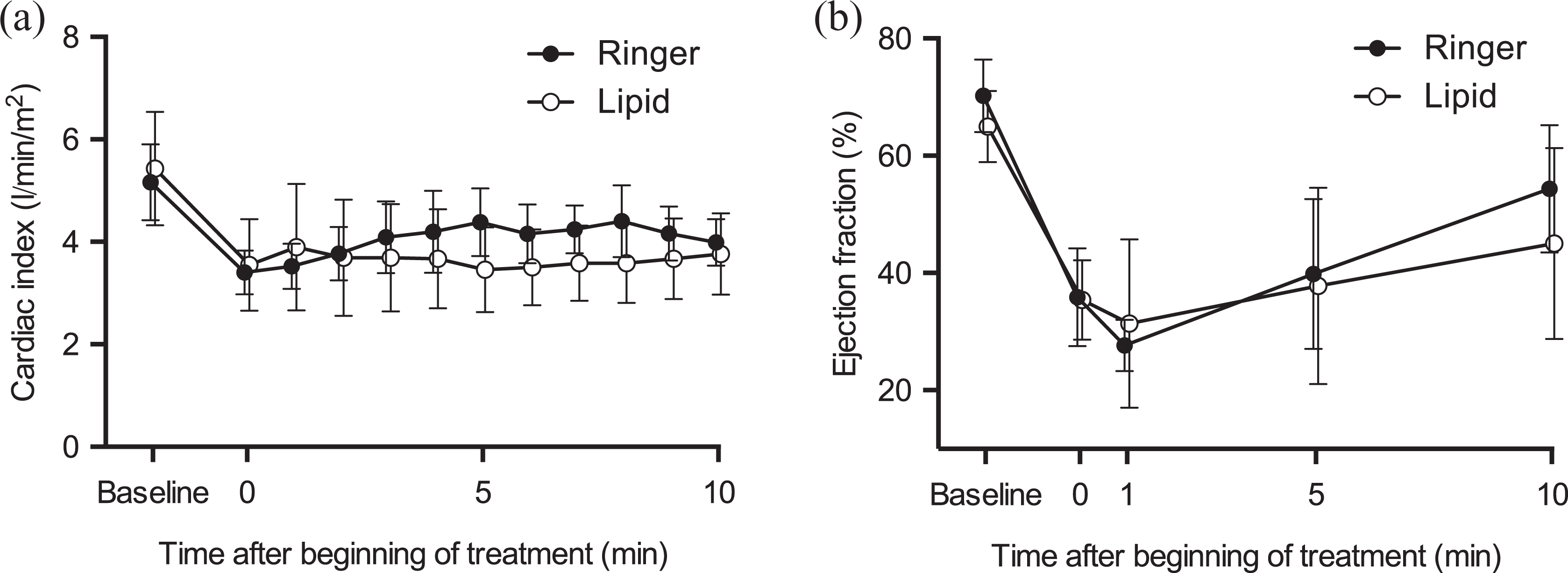
Cardiac index and left ventricular ER. After bupivacaine toxicity, lipid emulsion 4 mL/kg in 1 min did not affect the cardiac index (a) or the left ventricular ER (b). The interaction over time of mean cardiac index was significant (p = 0.01). In Sidak post-testing, there was, however, no difference at any time points between the groups. The interaction over time of the recovery of left ventricular ER was not statistically significant. EF: ejection fraction.
Mitochondrial respiration
During the different steps of the titration protocol, bupivacaine infusion decreased oxygen consumption on average by less than 10% as compared to baseline values; however, these differences did not reach statistical difference (p > 0.05). The mitochondrial complex I and II mediated oxygen flux was significantly higher (approximately 30%) after glutamate (p = 0.027), succinate (p = 0.029), un-coupler FCCP (p = 0.035), and rotenone (p = 0.044) in the Lipid group than in the Ringer group (Figure 5).

Cardiac mitochondrial respiration. Mean mitochondrial respiration was stimulated after administration of 4 mL/kg lipid emulsion in 1 min when compared to Ringer’s acetate through complex I after glutamate (Glu; p = 0.027), through complex I and II after succinate (Suc; p = 0.029), and after uncoupling, through complex I and II (FCCP; p = 0.035), and complex II alone after rotenone (Rot; p = 0.044). *p < 0.05; M: malate; Pal: palmitoyl-
Bupivacaine plasma concentrations
At the end of bupivacaine infusion, the mean (SD) peak bupivacaine concentration was 21.5 (2.5) mg/L in the Lipid group and 19.5 (2.9) mg/L in the Ringer group (p = 0.21; Figure 6). The mean (SD) AUC0–10 min of plasma total bupivacaine concentration was 105.2 (13.6) mg·min/L in the Lipid group and 88.1 (7.1) mg·min/L in the Ringer group (p = 0.019). In the Lipid group, the mean (SD) AUC0–10 min of plasma un-entrapped bupivacaine, 97.0 (14.5) mg·min/L, was 8% smaller than the AUC0–10 min of total bupivacaine concentration (p < 0.0001) but did not differ from the plasma concentration in the Ringer group (p = 0.20).

Bupivacaine plasma concentrations. When bupivacaine toxicity was treated with 4 mL/kg of either lipid emulsion or Ringer’s acetate, the mean area under the concentration–time curve of plasma total bupivacaine concentration from 0 min to 10 min was higher in the Lipid group than in the Ringer group (p = 0.019). The mean area under the concentration-time curve from 0 min to 10 min of un-entrapped (nonlipid-bound) bupivacaine was smaller than that of total concentration in the Lipid group (p < 0.0001) but did not differ from that of the total concentration in the Ringer group (p = 0.20).
The mean (SD) bupivacaine concentration in the tissue samples of the heart taken after the 10-min biopsy was 10.2 (6.2) mg/kg in the Lipid group and 12.0 (4.6) mg/kg in the Ringer group (p = 0.56). The corresponding lung concentrations were 22.5 (3.0) mg/kg and 19.7 (7.2) mg/kg, respectively (p = 0.38).
Discussion
In this model of bupivacaine-induced cardiotoxicity, the i.v. administration of a lipid emulsion dose 2.7 times greater per body weight than that clinically recommended expedited hemodynamic recovery in comparison with Ringer solution. The hemodynamic benefit was apparently due to peripheral vasoconstriction. Lipid emulsion stimulated myocardial mitochondrial respiration through complex I and II, but there were no signs of an inotropic effect of the lipid emulsion. There was no difference in left ventricular EF or in cardiac index between treatment with lipid emulsion and Ringer solution. Although statistically significant in comparison with Ringer treatment, the entrapment of bupivacaine by lipid emulsion in plasma was only modest (8%), and no significant effect on bupivacaine tissue concentrations was observed. Thus, bupivacaine entrapment seems unlikely to have significantly contributed to the hemodynamic recovery.
Hemodynamic recovery
We based our 4 mL/kg lipid emulsion dose on previously published rat 11 and pig models, 17 in which lipid emulsion was shown to increase MAP after bupivacaine intoxication. This dose of lipid emulsion was shown to increase MAP also without bupivacaine toxicity in our pilot study. In our main study, lipid emulsion did not increase cardiac index or left ventricular EF but clearly increased systemic arterial pressure by increasing systemic vascular resistance. The reason for the increased vascular tone, and the increased arterial pressure, is likely to be mediated by α1-adrenergic receptor activation as even a low dose of lipid emulsion has been shown to increase the reactivity of α1-adrenoceptors in human volunteers. 23,24 Lipid emulsion infused slowly together with heparin (activator of lipoprotein lipase) in human volunteers was found to increase systemic vascular resistance 2 h after the start of lipid infusion, 25 and in rat aortic rings, a vasoconstrictive effect of lipid emulsion was demonstrated after levobupivacaine-induced vasodilation. 26 This increased α1-adrenoceptor reactivity might also explain the cases of recovery and increased blood pressure with or without lipid emulsion administration in local anesthetic toxicity following adrenoceptor agonist administration. 27 –29
The α1-adrenoceptor of the vascular smooth muscle cells is a G-protein-coupled receptor whose activation leads to stimulation of phospholipase C activity and further, to activation of protein kinase C and eventually to vasoconstriction. 30 Protein kinase C can also be directly activated by unsaturated fatty acids, such as oleic acid, a component of Intralipid. 31 Another possibility of vasoconstrictive action of fatty acids is an effect on the endothelial nitric oxide synthase (eNOS) that produces vasodilatory nitric oxide. Free fatty acids included in Intralipid, for example, oleic and palmitic acid, inhibit eNOS independently of protein kinase C. 32,33 A direct activation of protein kinase C in smooth muscle and inhibition of eNOS in the endothelium of peripheral vessels may explain the rapidly increased peripheral vascular resistance after administration of lipid emulsion in our present study.
Mitochondrial respiration
Under normal conditions, the postnatal heart utilizes mainly fatty acids (about 70%) for the generation of ATP with glucose and lactate contributing to the rest. The main metabolic pathways include β-oxidation of fatty acids in the mitochondria and cytosolic glycolysis of glucose followed by intra-mitochondrial oxidation of pyruvate. Failure in cardiac bioenergetics causes mechanical failure of the heart. 34 Previous in vitro studies using either cardiomyocyte cell cultures or isolated cardiac mitochondria have revealed that bupivacaine dose-dependently decreased mitochondrial lipid-based respiration, whereas nonlipid-based mitochondrial respiration was not influenced by bupivacaine. 35 We therefore used a fatty acid substrate-uncoupler-inhibitor titration protocol in the present study to assess the effect of bupivacaine on cardiac mitochondrial respiration. 21 We also utilized a standardized approach to prepare high-quality tissue homogenate with high reproducibility of mitochondrial yield and mitochondrial function. 20,36 This technology offers some advantage over the use of isolated mitochondria; less tissue is required and the technology thus allows the use of tissue needle biopsies, the preparation is faster without a need for serial centrifugation steps, and finally, the possible drug effects on mitochondria are more likely to be present at the time of analysis as there is no risk of drug washout during tissue processing.
Interestingly, the oxygen consumption and lipid-based mitochondrial respiration remained virtually unchanged after bupivacaine toxicity, which contradicts previous findings of marked respiratory inhibition in isolated mitochondria. 14,15 The myocardial ATP production inhibition observed in isolated mitochondria required relatively high local anesthetic concentrations, and for instance, the half-inhibitory bupivacaine concentration for complex I was 0.38 mM, that is, 110 mg/L. 14 In the present study, the highest plasma concentration of bupivacaine reached was approximately 20 mg/L, and the mitochondria are likely exposed to even lower concentrations inside cardiomyocytes. The complex I mediated myocardial mitochondrial respiration, analyzed in isolated mitochondria, was not depressed by bupivacaine when asystole was induced by 10 mg/kg bupivacaine i.v. in rats. 16 Taken together, our findings with a nonisolation method suggest that the suppression of mitochondrial respiration by bupivacaine does not markedly account for bupivacaine cardiac toxicity at clinically relevant toxicity concentrations.
In the present study, the recovery effect of lipid emulsion was associated with a modest enhancement of mitochondrial respiration. By administering lipid emulsion, we demonstrated a slight increase in mitochondrial respiration through complexes I and II during both oxidative phosphorylation and after uncoupling of the electron transfer system from oxidative phosphorylation when compared to the lowest levels of respiration at the end of bupivacaine infusion. In the aforementioned study with isolated mitochondria after bupivacaine toxicity in rats, 16 such increased respiration through complex I could not be demonstrated. One possible reason for our contradicting findings is that in the rat study, the mitochondria were isolated after the bupivacaine injection so the respiration rates may not reflect the actual in vivo situation anymore after the isolation process. Also, no substrates of fatty acid oxidation were included in the rat mitochondrial respirometry study; the improved respiration in the present study may, at least partly, reflect the improved β-oxidation. This suggests that it is possible to slightly improve cardiac mitochondrial function by increasing fatty acid substrates. This improved respiration was not, however, accompanied by improved cardiac function, as both cardiac index and EF remained below the baseline level at the same time point and without differences between groups.
Entrapment of bupivacaine
In our previous studies in pigs and humans 8,37 –39 with the internationally recommended clinical rescue dose of lipid emulsion, 3 we could not demonstrate any relevant entrapment of bupivacaine or other local anesthetics in the plasma lipid phase. In the present study with the high 4 mL/kg bolus dose of lipid emulsion, the un-entrapped bupivacaine concentration, measured in the plasma aqueous phase, was about 8% lower than its total concentration. These results indicate that by increasing the amount of lipid, it is possible to entrap bupivacaine and increase the total amount of local anesthetic in circulation. A similar effect was seen when rats were administered a high dose (10 mL/kg) of lipid emulsion IV, and the total concentration of bupivacaine in whole blood even increased. 40 After much higher doses (total of 15 mL/kg of 30% lipid emulsion), it also seems possible to affect the tissue distribution of bupivacaine in rats. 9 In the present study, we did not detect any difference in tissue concentrations.
It is questionable if the observed slight entrapment of bupivacaine can contribute to clinical recovery. In our earlier study with the 100 times more lipophilic tricyclic antidepressant amitriptyline, 41 up to 55% of amitriptyline was entrapped by lipid emulsion without any effect on hemodynamic recovery in pigs. On the other hand, when the 10,000 times more lipophilic anti-arrhythmic amiodarone was entrapped almost entirely (up to 90%) into the plasma lipid phase, its hypotensive effect was prevented. 42 Thus, it seems likely that the degree of entrapment would need to be much higher than in the present study to contribute to hemodynamic effects.
Limitations
The cardiac biopsies were taken from the beating heart, which could affect the cardiac function during the experiment. A similar technique was, however, used in all pigs. The same model of biopsy needle is used clinically for biopsies of beating heart during heart transplantation. Also, the open chest has probably caused some degree of atelectasis in the lungs and a slight ventilation/perfusion mismatch. The degree of atelectasis, however, was minimized by adding positive end-expiratory airway pressure before performing thoracotomy.
A possible influence of isoflurane anesthesia on the results of mitochondrial function in the pigs cannot be fully excluded. Concentrations of isoflurane corresponding to about twice those used by us have slightly inhibited NADH oxidation (complex I) in isolated submitochondrial particles from pig heart. 43 However, there were no obvious indications of a toxic interaction on the mitochondrial respiratory chain level between 2% (end-tidal concentration) isoflurane and bupivacaine in our two study groups, which were treated identically until the start of rescue treatment. The well-known cardioprotective action of isoflurane, seen also with low anesthetic isoflurane concentrations, 44 seems not to be associated with disturbed mitochondrial respiration. 45
Some researchers have deemed the pig unsuitable for lipid rescue research, 46 because pigs may develop adverse reactions after high dose of lipid emulsion, 47 and rescue results in pig studies have been negative. 37 In the present study, administration of lipid emulsion only occasionally caused mild redness of the skin, but these reactions were not related to adverse hemodynamic effects, but rather to an enhanced recovery after a relatively high dose of lipid emulsion. 11,48 This suggests that a higher dose of lipid emulsion than recommended 3 is needed for the cardiovascular recovery.
Conclusions
Our study revealed at least three rescue mechanisms of action of minor or moderate power when 4 mL/kg of 20% lipid emulsion was given i.v. after bupivacaine-induced toxicity. First, lipid emulsion improved hemodynamic recovery by increasing systemic vascular resistance but did not improve cardiac index or EF, however. Second, bupivacaine did not markedly depress mitochondrial respiration at clinically relevant toxicity concentrations, but respiration through complex I and II was slightly stimulated with the administration of lipid emulsion. Third, the higher than recommended lipid emulsion dose also entrapped some of the bupivacaine in plasma but the amount was so small that it unlikely had any influence on the hemodynamic recovery.
Footnotes
Acknowledgments
We deeply thank Dr Maija Kaukonen, MD, PhD, for providing us with the PiCCO® PulsionFlex® monitor (funded by Academy of Finland) for the study. We gratefully acknowledge the work of Nada Bechara-Hirvonen for assaying the mitochondrial respiration. We thank Veikko Huusko and Olli Valtanen for their technical assistance. We also thank Jouko Laitila and Lisbet Partanen for the quantification of bupivacaine concentrations. We also appreciate the valuable comments by Professor James E. Heavner, PhD, during the preparation of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by research grants from Finska Läkaresällskapet, Finland, and Liv och Hälsa foundation, Finland.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.