
Abstract
C1q/TNF-related protein 12 (CTRP12) has been reported to play a key role in coronary artery disease. However, whether CTRP12 plays a role in the regulation of myocardial ischemia-reperfusion injury is not fully understood. The goals of this work were to assess the possible relationship between CTRP12 and myocardial ischemia-reperfusion injury. Here, we exposed cardiomyocytes to hypoxia/re-oxygenation (H/R) to establish an in vitro cardiomyocyte injury model of myocardial ischemia-reperfusion injury. Our results showed that H/R treatment resulted in a decrease in CTRP12 expression in cardiomyocytes. The up-regulation of CTRP12 ameliorated H/R-induced cardiomyocyte injury via the down-regulation of apoptosis, oxidative stress, and inflammation. In contrast, the knockdown of CTRP12 enhanced cardiomyocyte sensitivity to H/R-induced cardiomyocyte injury. Further investigation showed that CTRP12 enhanced the levels of nuclear Nrf2 and increased the expression of Nrf2 target genes in cardiomyocytes exposed to H/R. However, the inhibition of Nrf2 markedly diminished CTRP12-overexpression-mediated cardioprotective effects against H/R injury. Overall, these data indicate that CTRP12 protects against H/R-induced cardiomyocyte injury by inhibiting apoptosis, oxidative stress, and inflammation via the enhancement of Nrf2 signaling. This work suggests a potential role of CTRP12 in myocardial ischemia-reperfusion injury and proposes it as an attractive target for cardioprotection.
Introduction
Myocardial infarction is a fatal disease that has continued to be a leading cause of death and disability in recent years. 1 Coronary artery blockade leads to an insufficient blood supply to the heart, which causes myocardial ischemia. 2 Myocardial ischemia results in irreversible cardiac injury and dysfunction. However, rapid blood reperfusion can cause further harm to the myocardium, a clinical phenomenon called myocardial ischemia-reperfusion injury. 3 Various pathological changes occur during the progression of myocardial ischemia-reperfusion injury; however, the underlying mechanisms remain elusive. 4 Increased cardiac apoptosis, the excessive production of reactive oxygen species (ROS), and the extensive release of pro-inflammatory cytokines are closely related to cardiac injury and dysfunction during the development of myocardial ischemia-reperfusion injury. 5 Therefore, the identification of key genes that contribute to the modulation of cardiac apoptosis, oxidative stress, and inflammation underlying myocardial ischemia-reperfusion injury will help to provide novel and potential therapeutic targets for this disease.
C1q/TNF-related protein 12 (CTRP12) is a member of the CTRP super family that plays a key role in various physiological and pathological processes. 6 CTRP12 is widely expressed in numerous tissues and especially enriched in adipose tissue. 7 CTRP12 is identified as a novel adipokine that participates in the development of multiple diseases. The deletion of CTRP12 exacerbates insulin resistance and promotes the development of obesity in male mice consuming a high-fat diet. 8 The overexpression of CTRP12 improves insulin sensitivity and glucose metabolism in diabetic mice. 9 CTRP12 also plays a vital role in regulating lipid metabolism via the inhibition of triglyceride synthesis and export. 10 Notably, CTRP12 has been reported to have cardioprotective effects and play a vital role in coronary artery disease.6,11,12
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription factor that participates in the regulation of various biological processes under diverse pathological processes.13–15 When Nrf2 is activated, it enters the nucleus and binds to anti-oxidant response element (ARE), an enhancer region in its gene promoter, to trigger the expression of Nrf2 target genes. 16 The target genes of Nrf2 include antioxidant enzymes, molecular chaperones, and anti-inflammatory genes; thereby, Nrf2 has an outstanding cytoprotective role. 17 Notably, Nf2 has been proposed as a key a pivotal regulator in myocardial ischemia-reperfusion injury. 18 Activation of Nrf2 relieves the apoptosis, oxidative stress, and inflammation induced by myocardial ischemia-reperfusion injury.19,20 Therefore, Nrf2 is a promising target for the treatment of myocardial ischemia-reperfusion injury. The activation of Nf2 is modulated by multiple factors and mechanisms. A better understanding of the regulation of Nrf2 activation will provide novel insights into the development of Nrf2-related therapy for myocardial ischemia-reperfusion injury.
CTRP12 plays a key role in the regulation of cell apoptosis, oxidative stress, and inflammation.11,12 However, whether CTRP12 plays a role in the modulation of myocardial ischemia-reperfusion injury-related cardiac apoptosis, oxidative stress, and inflammation has not been fully studied. In this work, we aimed to determine the possible relationship between CTRP12 and myocardial ischemia-reperfusion injury using hypoxia/re-oxygenation (H/R)-induced cardiac injury in vitro.
Materials and methods
Cardiomyocytes
Primary cardiomyocytes were isolated from neonatal mice in compliance with previously described methods.21,22 Primary cardiomyocytes were maintained in specialized culture medium for cardiomyocytes (Procell, Wuhan, China) and placed in an incubator filled with a 5% CO2-containing atmosphere at 37°C. The characterization of primary cardiomyocytes was shown in Supplementary Figure S1. The mice were provided by the Medical Laboratory Animal Center of Xi’an Jiaotong University Health Science Center (Xi’an, China). The use of animals was approved by the Ethics Committee of Xi’an Jiaotong University Health Science Center (No: 2019-1267).
Establishment of H/R injury in cardiomyocytes
The establishment of H/R injury in cardiomyocytes was performed in accordance with a previously described method. 23 In brief, cardiomyocytes were placed into a tri-gas incubator filled with 1% O2/5% CO2/94% N2 (hypoxic conditions) and maintained for a period of 6 h. Afterwards, the cardiomyocytes were transferred to a normoxic incubator filled with 5% CO2/5% air (normoxic conditions) to achieve 12 h re-oxygenation.
Real-time quantitative PCR
The RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) was applied to isolate total RNA from cardiomyocytes. The QuantiTect Reverse Transcription Kit (Qiagen) was used to produce cDNA using total RNA as the template. Levels of cDNA were measured via real-time quantitative PCR (RT-qPCR) using the QuantiTect SYBR Green PCR Kit (Qiagen). RT-qPCR yield data were assessed via the 2ΔΔCt equation using β-actin for normalization.
Western blot
Protein exacts were prepared by lysing cardiomyocytes in RIPA buffer. After the determination of protein concentration, equivalent amounts of proteins were added to each well of the sodium dodecyl sulfate–polyacrylamide gels. After separation by electrophoresis, proteins were transferred to polyvinylidene fluoride membranes via electrophoretic transfer. Primary antibody incubation was performed after membrane blocking. Then, secondary antibody incubation was carried out, followed by color development with ECL western blot substrate. In the experiments, primary antibodies against CTRP12 (Bioss Antibodies, Beijing, China), Nrf2 (Abcam, Cambridge, UK), β-actin (Abcam), or Histone H3 (Abcam) were applied.
Cell transfection
CTRP12 siRNAs were designed and synthesized via GenePharma (Shanghai, China). CTRP12 vectors were constructed by subcloning the coding sequences of CTRP12 cDNA into the pcDNA3.1 expression vector. Cardiomyocytes were transfected with the siRNAs or vectors using Lipofectamine 3000 Transfection Reagent (Thermo Fisher Scientific, Waltham, MA, USA) in accordance with the manufacturers’ instructions.
Cell viability assay
Cardiomyocytes were plated into a 96-well plate and transfected with CTRP12 siRNAs or vectors for 48 h prior to H/R exposure. To monitor cell viability, 10 µl of MTT stock solution was added to each well, and cells were further cultured for 4 h. Then, a solubilizing agent was added to dissolve the formazan crystals. A microplate reader (Bio-Rad, Hercules, CA, USA) was employed to measure the optical density value of the colorimetric solution at 570 nm.
Cell apoptosis assay
Cardiomyocytes were dissociated with trypsin and washed with phosphate buffered saline (PBS). Following cell enumeration, 1 × 105 cells were collected and incubated with Annexin V-FITC/PI Staining Solution (Beyotime, Shanghai, China). Following incubation for 20 min in the dark at room temperature, cell apoptosis was evaluated via flow cytometry.
ROS assay
Levels of ROS were monitored using an ROS-sensitive probe, 2′,7′-dichlorofluorescin diacetate (DCFDA) (Abcam, Cambridge, UK). Cells were collected and re-suspended in 1X Buffer containing 20 μM of DCFDA, followed by incubation for 45 min at 37°C. The fluorescence intensity was measured using a Varioskan Flash Multimode Reader (Thermo Fisher Scientific, Waltham, MA, USA) at excitation and emission wavelengths of 485 and 535 nm, respectively.
Quantification of inflammatory cytokines
The concentrations of tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6 in culture media was assessed using ELISA kits (R&D Systems China, Shanghai, China) in accordance with standard procedures.
Luciferase reporter assay
The Nrf2 reporter vector, pGL4.37[luc2P/ARE/Hygro] (Promega, Madison, WI, USA), containing ARE sites, was utilized to monitor the transcriptional activity of Nrf2. In general, cardiomyocytes were co-transfected with the Nrf2 reporter vector, the control Renilla luciferase vector and CTRP12 siRNAs or vectors for 48 h prior to H/R treatment. Then, luciferase activity within cells was quantified in compliance with the protocols of the Dual-Glo Luciferase Assay System (Promega).
Statistical analysis
Quantitative data are expressed as means ± standard deviation. Data were analyzed with GraphPad Prism version 8.0 software (GraphPad Software, San Diego, CA, USA). Differences between two groups were assessed via Student’s t-test, whilst differences between three or more groups were assessed via one-way analysis of variance followed by Tukey’s post hoc test. A P value less than 0.05 denoted a statistically significant difference. All experiments were repeated at least three times to ensure the reproducibility of the data.
Results
CTRP12 expression is decreased in cardiomyocytes exposed to H/R
To determine the possible association between CTRP12 and H/R-induced cardiomyocyte injury, we first examined alterations in the expression of CTRP12 in cardiomyocytes in response to H/R treatment. We found that mRNA levels of CTRP12 were markedly decreased in cardiomyocytes exposed to H/R compared with those in normal cells (Figure 1(A)). Moreover, protein levels of CTRP12 were also decreased by H/R treatment in cardiomyocytes (Figure 1(B) and (C)). Overall, these data suggest that CTRP12 expression is decreased in cardiomyocytes exposed to H/R.

Effect of H/R treatment on CTRP12 expression in cardiomyocytes. (A) Levels of CTRP12 mRNA were examined via RT-qPCR. (B, C) Levels of CTRP12 protein were determined via western blotting (N = 3, **p < 0.01).
Up-regulation of CTRP12 attenuated H/R-induced apoptosis and oxidative stress in cardiomyocytes
To explore the function of CTRP12 in regulating H/R-induced cardiomyocyte injury, we used the CTRP12 expression vector to overexpress CTRP12. The levels of CTRP12 expression were much higher in CTRP2-vector-transfected cells than that in empty-vector-transfected cells (Figure 2(A) to (C)). The viability of cardiomyocytes was markedly impaired by H/R exposure, which was effectively rescued by CTRP12 overexpression (Figure 2(D)). Apoptosis in cardiomyocytes evoked by H/R was markedly attenuated by CTRP12 overexpression (Figure 2(E) and (F)). Moreover, the up-regulation of CTRP12 prevented the production of ROS in cardiomyocytes exposed to H/R (Figure 2(G)). Collectively, these data indicate that the up-regulation of CTRP12 ameliorates H/R-induced apoptosis and oxidative stress in cardiomyocytes.
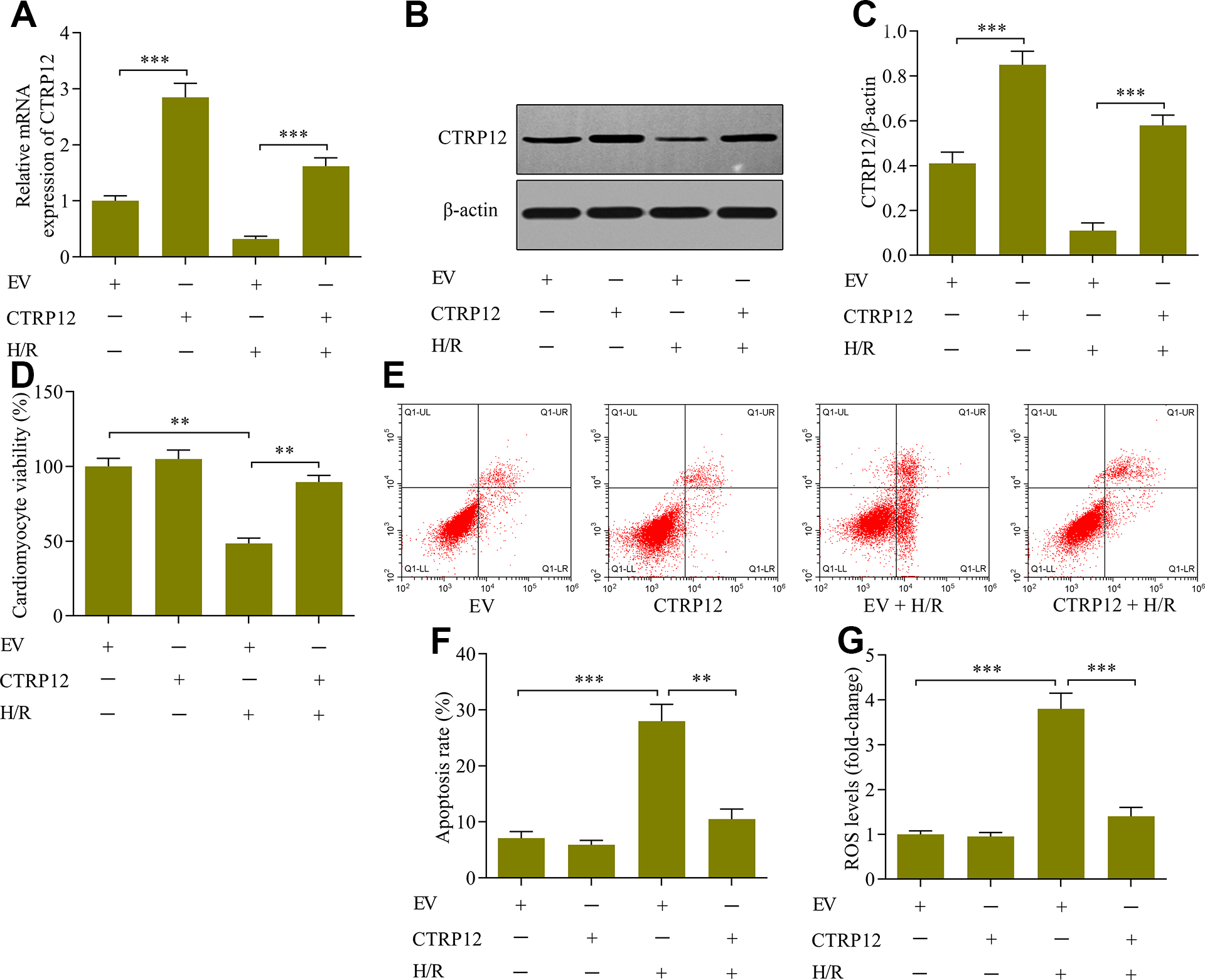
Effect of CTRP12 overexpression on H/R-induced apoptosis and oxidative stress in cardiomyocytes. Cardiomyocytes were transfected with CTRP12 vectors or empty vectors (EV) and cultured for 48 h before H/R was performed. Levels of CTRP12 (A) mRNA or (B, C) protein were detected via RT-qPCR or western blotting, respectively (N = 3, ***p < 0.001). (D) Cardiomyocyte viability was measured via MTT assay (N = 4, **p < 0.01). (E, F) Cardiomyocyte apoptosis was assessed via Annexin V-FITC/PI apoptotic assay (N = 3, **p < 0.01and ***p < 0.001). EV [viable cells (92 ± 2.2)%; early apoptotic cell (3.5 ± 0.6)%; late apoptotic cells (3.2 ± 0.5)%]; CTRP12 [viable cells (94 ± 2.5)%; early apoptotic cell (2.4 ± 0.4)%); late apoptotic cells (3.6 ± 0.8)%]; EV + H/R [viable cells (70.5 ± 4.2)%; early apoptotic cell (19.2 ± 2.6)%; late apoptotic cells (8.4 ± 1.5)%]; EV + H/R [viable cells (70.5 ± 4.2)%; early apoptotic cell (19.2 ± 2.6)%; late apoptotic cells (8.4 ± 1.5)%]; CTRP12 + H/R [viable cells (89.5 ± 5.2)%; early apoptotic cell (4.2 ± 0.6)%; late apoptotic cells (6.8 ± 1.1)%]. (G) ROS levels were monitored via ROS assay (N = 3, ***p < 0.001).
Knockdown of CTRP12 exacerbated H/R-induced apoptosis and oxidative stress in cardiomyocytes
To confirm that CTRP12 is involved in regulation of H/R-induced cardiomyocyte apoptosis and oxidative stress, we further performed loss-of-function experiments. We used CTRP12 siRNA to deplete the expression of CTRP12 in cardiomyocytes (Figure 3(A) and (B)). Depletion of CTRP12 further decreased the viability of cardiomyocytes exposed to H/R (Figure 3(C)). Moreover, knockdown of CTRP12 exacerbated H/R-induced apoptosis (Figure 3(D) and (E)) and ROS production (Figure 3F) in cardiomyocytes. In summary, these findings suggest that the loss of CTRP12 enhances cardiomyocyte sensitivity to H/R-induced apoptosis and oxidative stress.

Effect of CTRP12 depletion on H/R-induced apoptosis and oxidative stress in cardiomyocytes. Cardiomyocytes were transfected with CTRP12 siRNA or negative control (NC) siRNA and cultured for 48 h before H/R was performed. (A, B) Levels of CTRP12 protein were detected via western blotting (N = 3, ***p < 0.001). (C) Cardiomyocyte viability was assessed via MTT assay (N = 4, **p < 0.01). (D, E) Cardiomyocyte apoptosis was evaluated via Annexin V-FITC/PI apoptotic assay (N = 3, **p < 0.01 and ***p < 0.001). NC siRNA [viable cells (93.5 ± 2.5)%; early apoptotic cell (2.5 ± 0.5)%; late apoptotic cells (3.4 ± 0.7)%]; CTRP12 siRNA [viable cells (93.5 ± 1.5)%; early apoptotic cell (2.2 ± 0.4)%; late apoptotic cells (3.9 ± 0.5)%]; NC siRNA + H/R [viable cells (75.2 ± 4.5)%; early apoptotic cell (6.5 ± 0.8)%; late apoptotic cells (16.4 ± 1.2)%]; CTRP12 siRNA + H/R [viable cells (59.2 ± 5.2)%; early apoptotic cell (28.5 ± 2.9)%; late apoptotic cells (10.4 ± 1.8)%]. (F) ROS levels were examined via ROS assay (N = 3, **p < 0.01 and ***p < 0.001).
CTRP12 modulates the inflammatory response induced by H/R in cardiomyocytes
To further explore the role of CTRP12 in regulating H/R-induced cardiomyocyte injury, we investigated the effect of CTRP12 on regulating the inflammatory response induced by H/R in cardiomyocytes. The levels of the pro-inflammatory cytokines TNF-α, IL-1β, and IL-6 were markedly elevated by H/R exposure in cardiomyocytes (Figure 4(A) to (C)). Notably, the up-regulation of CTRP12 markedly inhibited the release of TNF-α, IL-1β, and IL-6 in cardiomyocytes exposed to H/R (Figure 4(A) to (C)). In contrast, the loss of CTRP12 enhanced the release of TNF-α, IL-1β, and IL-6 in cardiomyocytes exposed to H/R (Figure 4(D) to (F)). Collectively, these results suggest that CTRP12 modulates the inflammatory response induced by H/R in cardiomyocytes.
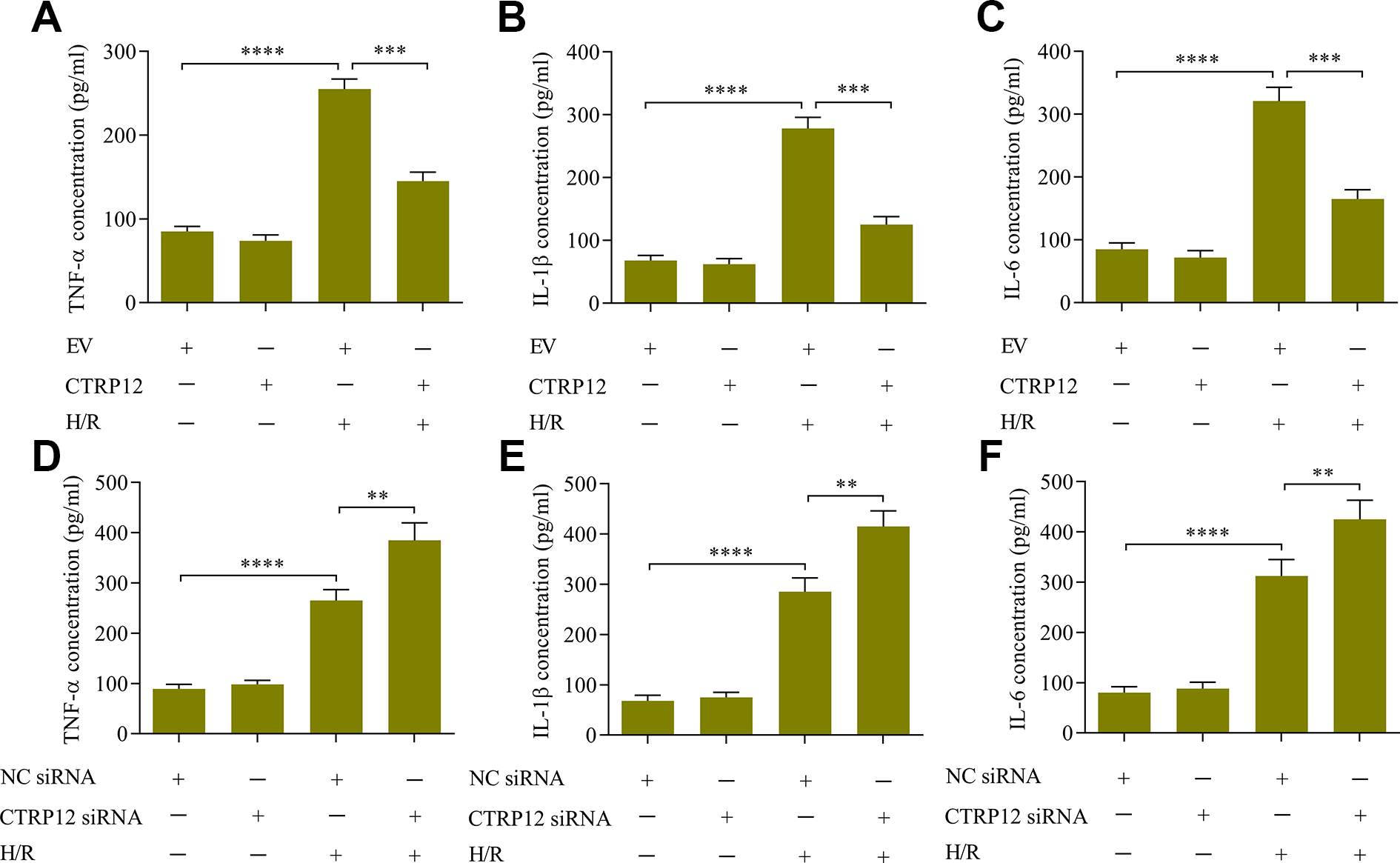
Effect of CTRP12 on the inflammatory response induced by H/R in cardiomyocytes. The effect of CTRP12 overexpression on the concentrations of (A) TNF-α, (B) IL-1β and (C) IL-6 was measured via ELISA (N = 4, ***p < 0.001, and ****p < 0.0001). The effect of CTRP12 depletion on the concentration of (D) TNF-α, (E) IL-1β, and (F) IL-6 was quantified via ELISA (N = 4, **p < 0.01 and ****p < 0.0001).
CTRP12 contributes to the modulation of Nrf2 activation in cardiomyocytes exposed to H/R
To dissect the mechanism by which CTRP12 modulates H/R-induced cardiomyocyte injury, we investigated the effect of CTRP12 on Nrf2 signaling. The results showed that the up-regulation of CTRP12 significantly increased the levels of nuclear Nrf2 (Figure 5(A) and (B)) in cardiomyocytes exposed to H/R. Moreover, the overexpression of CTRP12 enhanced the transcriptional activity of Nrf2/ARE (Figure 5(C)) and promoted the expression of the Nrf2 target genes HO-1 and NQO-1 (Figure 5(D) and (E)) in cardiomyocytes exposed to H/R. In contrast, the loss of CTRP12 impeded the activation of Nrf2 signaling in cardiomyocytes exposed to H/R (Figure 5(F) to (H)). In short, these results confirm that CTRP12 enhances Nrf2 activation in cardiomyocytes exposed to H/R.
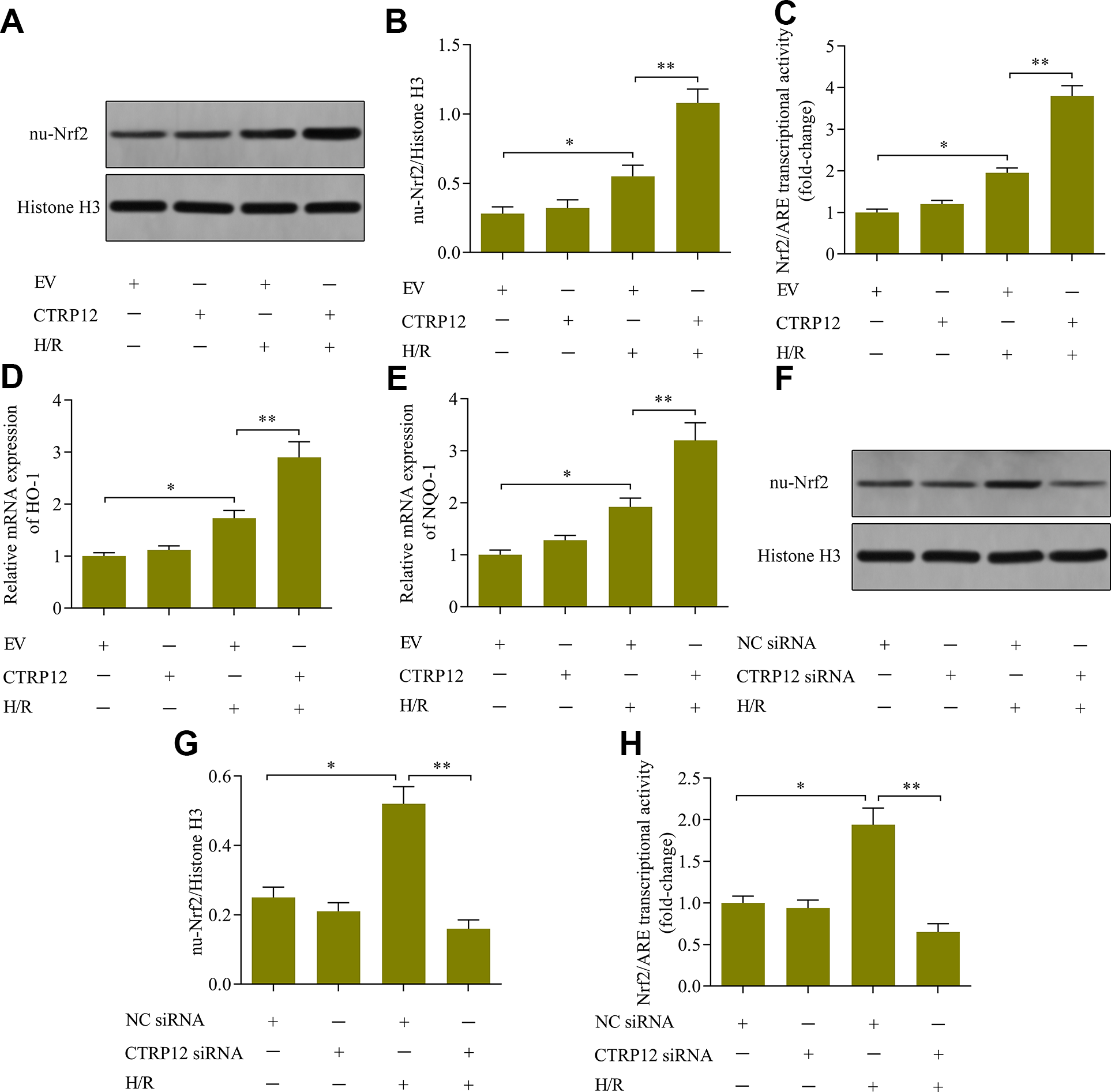
Effect of CTRP12 on Nrf2 signaling in cardiomyocytes exposed to H/R. (A, B) The effect of CTRP12 overexpression on levels of nuclear Nrf2 was determined via western blotting (N = 3, *p < 0.05 and **p < 0.01). (C) The effect of CTRP12 overexpression on Nrf2/ARE transcriptional activity was monitored via luciferase reporter assay (N = 4, *p < 0.05 and **p < 0.01). The effect of CTRP12 on the mRNA levels of (D) HO-1 and (E) NQO-1 was examined via RT-qPCR (N = 3, *p < 0.05 and **p < 0.01). (F, G) The effect of CTRP12 knockdown on the levels of nuclear Nrf2 were determined via western blotting (N = 3, *p < 0.05 and **p < 0.01). (H) The effect of CTRP12 knockdown Nrf2/ARE transcriptional activity was assessed via luciferase reporter assay (N = 3, *p < 0.05 and **p < 0.01).
Inhibition of Nrf2 diminished CTRP12-mediated cardioprotective effects
To validate whether CTRP12 exerts cardioprotective effects via Nrf2, we investigated the effect of Nrf2 inhibition on CTRP12-overexpression-mediated effects in cardiomyocytes exposed to H/R. We used the Nrf2 inhibitor ML385 to inhibit the activity of Nrf2 (Figure 6(A)). Treatment with ML385 markedly weakened Nrf2 activity in cardiomyocytes overexpressing CTRP12 (Figure 6(A)). As expected, the inhibitory effect of CTRP12 overexpression on H/R-induced apoptosis (Figure 6(B) and (C)), oxidative stress (Figure 6(D)), and the inflammatory response (Figure 6(E) to (G)) in cardiomyocytes was partially reversed by Nrf2 inhibition. In short, these findings confirm that CTRP12 confers cardioprotective effects via Nrf2.

Effect of Nrf2 inhibition on CTRP12-mediated cardioprotective effects. Cardiomyocytes were transfected with CTRP12 vectors and cultured for 48 h with ML385 prior to H/R exposure. (A) Nrf2 transcriptional activity was monitored via luciferase reporter assay (N = 3, *p < 0.05 and **p < 0.01). (B, C) Cardiomyocyte apoptosis was measured via Annexin V-FITC/PI apoptotic assay (N = 3, **p < 0.01, and ***p < 0.0001). Control [viable cells (91.5 ± 2.8)%; early apoptotic cell (2.5 ± 0.4)%; late apoptotic cells (5.2 ± 0.9)%]; Vehicle + EV + H/R [viable cells (73.8 ± 3.2)%; early apoptotic cell (17.6 ± 1.8)%; late apoptotic cells (6.8 ± 1.2)%]; ML385 + EV + H/R [viable cells (57.2 ± 4.2)%; early apoptotic cell (29.5 ± 2.6)%; late apoptotic cells (11.5 ± 1.5)%]; Vehicle + CTRP12 + H/R [viable cells (88.5 ± 5.5)%; early apoptotic cell (4.2 ± 0.7)%; late apoptotic cells (6.4 ± 0.9)%]; ML385 + CTRP12 + H/R [viable cells (77.5 ± 3.7)%; early apoptotic cell (15.3 ± 1.2)%; late apoptotic cells (5.6 ± 0.8)%]. (D) ROS levels were assessed via ROS assay (N = 3, **p < 0.01, and ***p < 0.0001). The levels of (E) TNF-α, (F) IL-1β, and (G) IL-6 were quantified via ELISA (N = 4, **p < 0.01 and ***p < 0.0001).
Discussion
This work provides compelling evidence that CTRP12 exerts a key role in modulation of H/R-induced cardiac injury. We found that CTRP12 expression was decreased in cardiomyocytes exposed to H/R. Functional results demonstrated that the up-regulation of CTRP12 ameliorated cardiac apoptosis, ROS production, and pro-inflammatory cytokine release evoked by H/R, whilst the loss of CTRP12 enhanced cardiomyocyte sensitivity to H/R-induced cardiac injury. Further investigation determined that the cardioprotective role of CTRP12 was related to the activation of Nrf2 signaling (Figure 7). In short, our work highlights a new role of CTRP12 in regulating H/R-induced cardiac injury.

Schematic model illustrating the role of CTRP12-mediated Nrf2 signaling in the inhibition of H/R-induced apoptosis, oxidative stress, and the inflammatory response.
The down-regulation of CTRP12 is associated with the development of various diseases. The levels of CTRP12 are decreased in mice with insulin resistance and obesity.7,9 The reduced expression of CTRP12 is detected in patients with polycystic ovarian syndrome.24,25 Moreover, decreased serum levels of CTRP12 are also found in patients with coronary artery disease. 26 Reduced levels of CTRP12 are correlated with kidney dysfunction. 27 These studies indicate that CTRP12 is essential for maintaining normal functions, and its dysregulation may be correlated with disease development. In this study, we found that CTRP12 expression was decreased in cardiomyocytes in response to H/R treatment. Our results suggest that CTRP12 may be involved in regulating H/R-induced cardiac injury. Interestingly, a recent study reported that CTRP12 overexpression alleviates isoproterenol-induced cardiac fibrosis in mice, suggesting a critical role of CTRP12 in cardiac fibrosis. Considering that H/R-induced cardiac injury is an in vitro model of myocardial ischemia-reperfusion injury, CTRP12 may play a role in myocardial ischemia-reperfusion injury in vivo.
CTRP12 is essential for cell survival under adverse conditions. It is reported that the overexpression of CTRP12 prohibits the apoptosis of human umbilical vein endothelial cells under serum deprivation. 11 Notably, CTRP12 plays a key role in maintaining cardiomyocyte cell survival. The up-regulation of CTRP12 enables cardiomyocyte survival under lipopolysaccharide (LPS) stimulation and represses LPS-evoked cardiomyocyte apoptosis, 12 indicating an outstanding cardioprotective role of CTRP12. In line with these studies, our findings demonstrated that the up-regulation of CTRP12 increased cardiomyocyte survival under H/R exposure and reduced H/R-induced cardiomyocyte apoptosis. Therefore, our work confirms a cardioprotective role of CTRP12.
CTRP12 has been reported to exert a key role in modulating oxidative stress in cardiomyocytes. It is reported that the up-regulation of CTRP12 enhances the anti-oxidant capability of cardiomyocytes against LPS-induced oxidative stress by inhibiting the production of ROS and malondialdehyde, and increasing the activity of superoxide dismutase. 12 These findings suggest a potent antioxidant activity of CTRP12. Consistent with this hypothesis, the results of our work demonstrated that the up-regulation of CTRP12 markedly decreased H/R-induced production of ROS in cardiomyocytes. Thus, our work confirms that CTRP12 exerts an antioxidant role in cardiomyocytes.
CTRP12 acts as a potent anti-inflammatory adipokine. Low levels of serum CTRP12 in patients with coronary artery disease are correlated with high levels of TNF-α and IL-6. 26 The overexpression of CTRP12 suppresses the expression of pro-inflammatory genes in adipose tissues of obese mice. 7 In cultured macrophages, the overexpression of CTRP12 diminishes the release of pro-inflammatory cytokines evoked by LPS or TNF-α.7,11 Moreover, the up-regulation of CTRP12 repressed the release of TNF-α, IL-1β, and IL-6 induced by LPS in cardiomyocytes, suggesting a potential role of CTRP12 in regulating the inflammatory response of cardiomyocytes. 12 In line with these studies, our results demonstrated that CTRP12 overexpression significantly repressed the release of TNF-α, IL-1β, and IL-6 in cardiomyocytes following H/R exposure. Collectively, our study confirms a potent anti-inflammatory role of CTRP12 in cardiomyocytes.
Nrf2 is a remarkable cytoprotective protein that has anti-apoptotic, anti-oxidative stress, and anti-inflammatory roles.28–30 Mounting evidence shows that activation of Nrf2 protects against H/R-induced cardiac injury in vitro and myocardial ischemia-reperfusion injury in vivo.31–34 Various factors regulate Nrf2 activation during H/R-induced cardiac injury; however, the precise molecular mechanisms remain largely unknown. CTRP12 has been reported to exert a key role in the regulation of Nrf2 activation. It is reported that the up-regulation of CTRP12 enhances the nuclear translocation of Nrf2 and promotes the expression of Nrf2 target genes in LPS-stimulated cardiomyocytes. 12 However, whether CTRP12-mediated Nrf2 activation also exists in H/R-induced cardiac injury is unknown. Notably, our work found that the overexpression of CTRP12 potentiated the activation of Nrf2 in cardiomyocytes exposed to H/R injury, confirming a regulatory effect of CTRP12 in Nrf2 signaling. Moreover, our results verified that the inhibition of Nrf2 markedly abolished the CTRP12-overexpression-mediated cytoprotective effects in H/R-injured cardiomyocytes. Therefore, our work confirms that CTRP12 exerts its cardioprotective effects via the modulation of Nrf2 signaling. These findings suggest that CTRP12-mediated Nrf2 activation may be involved in the progression of myocardial ischemia-reperfusion injury. Considering that Nrf2 activation is protective against myocardial ischemia-reperfusion injury,35–37 targeting CTRP12 to activate Nrf2 may represent a potential treatment option for this disease.
In summary, the findings of this work demonstrate that CTRP12 is capable of suppressing apoptosis, oxidative stress, and inflammation in cardiomyocytes after exposure to H/R by enhancing the activation of Nrf2 signaling. Our work underlines a cardioprotective role of CTRP12 that may play a role in myocardial ischemia-reperfusion injury. CTRP12 may serve as a novel target for cardioprotection. However, the precise cardioprotective role of CTRP12 requires further validation in vivo using animal models.
Supplemental Material
Supplemental Material, sj-pdf-1-het-10.1177_09603271211021880 - Up-regulation of CTRP12 ameliorates hypoxia/re-oxygenation-induced cardiomyocyte injury by inhibiting apoptosis, oxidative stress, and inflammation via the enhancement of Nrf2 signaling
Supplemental Material, sj-pdf-1-het-10.1177_09603271211021880 for Up-regulation of CTRP12 ameliorates hypoxia/re-oxygenation-induced cardiomyocyte injury by inhibiting apoptosis, oxidative stress, and inflammation via the enhancement of Nrf2 signaling by Ai-Ping Jin, Qian-Rong Zhang, Cui-Ling Yang, Sha Ye, Hai-Juan Cheng and Yuan-Yuan Zheng in Human & Experimental Toxicology
Footnotes
Author contributions
A.-P.J. designed the study, performed the experiments and drafted the manuscript. Q.-R.Z. performed the experiments. C.-L.Y. collected and analyzed the data. S.Y. collected and analyzed the data. H.-J.C. collected and analyzed the data. Y.-Y.Z. collected and analyzed the data.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Key Research & Development Project of Shaanxi Province (2019SF-101).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.