
Abstract
Sphingomyelin synthase 2 (SMS2) is a vital contributor to tissue injury and affects various pathological processes. However, whether SMS2 participates in the modulation of cardiac injury in myocardial infarction has not been determined. This study aimed to evaluate the potential role of SMS2 in the regulation of cardiomyocyte injury induced by hypoxia, an in vitro model for studying myocardial infarction. Our data revealed that SMS2 expression was significantly upregulated in cardiomyocytes in response to hypoxia. Loss-of-function experiments revealed that knockdown of SMS2 markedly restored the viability of cardiomyocytes impaired by hypoxia, and attenuated hypoxia-evoked apoptosis and reactive oxygen species (ROS) generation. In contrast, cardiomyocytes that highly expressed SMS2 were more sensitive to hypoxia-induced injury. Moreover, SMS2 deficiency enhanced the activation of nuclear factor erythroid 2-related factor 2 (Nrf2) signaling through inactivation of glycogen synthase kinase-3β. Notably, suppression of Nrf2 markedly abrogated SMS2 knockdown-mediated cardioprotective effects on hypoxia-exposed cardiomyocytes. Our results illustrate that downregulation of SMS2 exerts a cardioprotective function by protecting cardiomyocytes from hypoxia-induced apoptosis and oxidative stress through enhancement of Nrf2 activation. Our study indicates a potential role of SMS2 in the modulation of cardiac injury, which may contribute to the progression of myocardial infarction.
Introduction
Myocardial infarction is a common manifestation of ischemic heart disease, which occurs as a result of coronary artery blockage, and leads to insufficient blood supply to the heart. 1 Myocardial infarction causes irreversible cardiac damage and dysfunction, and is considered as a major cause of disability and death. 2 Patients with myocardial infarction may suffer from several manifestations including chest pain, dyspnea, weakness, nausea, sweating or combined symptoms. 3,4 Energy supply disturbances, calcium overload, inflammatory response, and oxidative stress caused by myocardial infarction contribute to the injury of cardiomyocytes. 5,6 However, the detailed molecular mechanisms underlying myocardial infarction-induced cardiomyocyte injury remain elusive. A better understanding of the underlying mechanisms of myocardial infarction-induced cardiomyocyte injury will help exploit novel therapies for myocardial infarction.
Sphingomyelin synthase 2 (SMS2), a member of the sphingomyelin synthases family, plays a vital role in the modulation of sphingomyelin synthesis. 7 SMS2 is predominantly located at the cell membrane, and affects signal transduction and the physiological functioning of the cell. 8 SMS2 plays a critical role in the regulation of cell survival and death, and its dysregulation has been implicated in various human disorders. 9 SMS2 deficiency impedes the progression of atherosclerosis. 10 High levels of SMS2 expression promote lung injury by amplifying proinflammation. 11 SMS2 knockout promotes apoptosis in mossy cells in the hippocampus, and is caused by prenatal alcohol exposure. 12 Moreover, SMS2 is involved in oxidative stress-induced endothelial dysfunction. 13 Notably, a recent study reported that SMS2 inhibition ameliorates ischemia-induced brain injury, 14 indicating that SMS2 may participate in the regulation of ischemic injury.
Nuclear factor erythroid 2-related factor 2 (Nrf2) is principally responsible for modulating cellular antioxidant defense mechanisms. 15 Nrf2 is a nuclear transcription factor that initiates expression of cytoprotective genes in response to oxidative stress. Generally, Nrf2 is stabilized and translocated into the nucleus in response to oxidative stress. 16 In the nucleus, Nrf2 is capable of binding antioxidant response elements (ARE) within the promoters of Nrf2 target genes. 17 Activation of Nrf2/ARE signaling is modulated via multiple mechanisms. 18 Growing evidence indicates that glucogen synthase kinase (GSK)-3β acts as a key modulator of Nrf2. 19 –21 Reports indicate that activation of GSK-3β via dephosphorylating a Ser9 residue stimulates degradation of Nrf2 and blocks activation of Nrf2/ARE signaling. 20,21 Importantly, the GSK-3β/Nrf2/ARE axis reportedly plays a pivotal role in the regulation of myocardial ischemic injury. 22,23 Therefore, understanding the mechanisms involved in the modulation of the GSK-3β/Nrf2/ARE axis may provide novel insights regarding the development of effective cardioprotective therapy.
SMS2 has been reported to play a key role in regulating tissue injury via regulating cell survival and oxidative stress. However, its role in the modulation of myocardial infarction-induced injury remains unknown. This study was designed to evaluate the potential role of SMS2 in the regulation of cardiomyocyte injury induced by hypoxia. Our data revealed that SMS2 expression was significantly upregulated in cardiomyocytes in response to exposure to hypoxic conditions. Interestingly, knockdown of SMS2 markedly restored the viability of cardiomyocytes previously exposed to hypoxia, and attenuated hypoxia-evoked apoptosis and reactive oxygen species (ROS) generation. Further data indicated that SMS2 knockdown enhanced activation of Nrf2 via inactivation of GSK-3β. Notably, suppression of Nrf2 markedly abrogated SMS2 knockdown-mediated cardioprotective effects observed in hypoxia-exposed cardiomyocytes. Taken together, our results demonstrate that downregulation of SMS2 protects cardiomyocytes from hypoxia-induced apoptosis and oxidative stress by enhancing Nrf2 activation via inactivation of GSK-3β.
Materials and methods
Cardiomyocyte culture
Cultures of mouse cardiomyocytes were obtained from neonatal mice, as previously described. 24 Isolated cardiomyocytes were cultivated in mouse cardiomyocyte complete medium (Procell, Wuhan, China) and maintained in a 5% CO2-containing atmosphere at 37°C.
Hypoxia exposure of cardiomyocytes
Hypoxic injury of cardiomyocytes was established according to a previously described method. 25 In brief, cardiomyocytes were cultured in a gaseous mixture containing 94% N2/5% CO2/1% O2 for 24 h at 37°C. Cardiomyocytes cultured under normoxic conditions functioned as controls.
Real-time quantitative PCR (RT-qPCR)
To isolate and purify total RNA, cardiomyocytes were homogenized in lysis buffer provided within the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany). Total RNA was reverse-transcribed to form complementary DNA using a QuantiTect Reverse Transcription Kit (Qiagen) in accordance with the manufacturer’s instructions. RT-qPCR for quantification of complementary DNA levels was carried out with the QuantiTect SYBR Green PCR Kit (Qiagen). RT-qPCR data was analyzed via the comparative ΔΔCt method, using β-actin to normalize gene expression.
Western blot
Cardiomyocytes were lysed in Radio-Immuno-Precipitation Assay (RIPA) buffer for protein extraction. After quantification of protein levels, a total of 20 µg protein was collected and separated via sodium dodecyl sulfate polyacrylamide gel electrophoresis. Then, separated proteins were electro-transferred to polyvinylidene fluoride (PVDF) membranes. After transfer, PVDF membranes were immersed in blocking buffer prior to primary antibody incubation. Membranes were incubated with primary antibodies overnight at 4°C. Then, secondary antibodies conjugated to horseradish peroxidase and ECL western blot substrate were utilized to visualize protein bands on PVDF membranes. Primary antibodies against SMS2 (Invitrogen), Nrf2 (Abcam), β-actin (Abcam) or Histone H3 (Abcam) and phospho-GSK-3β (Ser9) (Cell Signaling Technology) were used.
Transfection of siRNAs and vectors
The sequences of siRNAs targeting SMS2 were designed and synthesized by GenePharma (Shanghai, China). Vectors expressing SMS2 were constructed by subcloning SMS2 coding sequences into pcDNA3.1 vectors. Lipofectamine 3000 Transfection Reagent (Thermo Fisher Scientific, Waltham, MA, USA) was used to transfect siRNAs or/and vectors into cardiomyocytes in accordance with the manufacturer’s instructions.
MTT cell viability assay
Cardiomyocytes were seeded into a ninety-six-well plate and cultivated overnight to allow cells to adhere to plate surfaces. Then, cultured cardiomyocytes were transfected with siRNAs or/and vectors and incubated for 48 h prior to hypoxia exposure. To determine cell viability, 10 µl per well of MTT solution (Thermo Fisher Scientific, Waltham, MA, USA) was added to cells and incubated 4 h at 37°C. Formazan crystals were dissolved with solubilizing agent and the optical density value of the colorimetric solution was assessed using a microplate reader (Bio-Rad, Hercules, CA, USA) at a wavelength of 570 nm.
Caspase-3 activity assay
Caspase-3 activity was assessed via a Caspase 3 Activity Assay Kit (Beyotime, Shanghai, China) in compliance with the manufacturer’s protocols. Briefly, cells lysed by lysis buffer and the supernatants were collected for detection. Supernatants were added to a new 96-well plate at 50 μl/well. Then, 10 µl of Ac-DEVD-pNA (2 mM) and 40 µl of reaction buffer were added to each well and incubated for 2 h at 37°C. The absorbance at 405 nm was measured with a microplate reader a microplate reader (Bio-Rad, Hercules, CA, USA).
Annexin V-FITC/PI apoptotic assay
When apoptosis was detected, cardiomyocytes were rinsed with phosphate buffered saline (PBS) and resuspended to obtain single-cell suspensions. Cardiomyocytes were counted, and 1 × 105 cells were incubated with annexin V-FITC/PI staining solution in annexin V Binding Buffer at room temperature in the dark. Then, apoptosis rate of cardiomyocytes was analyzed using flow cytometry.
Measurement of oxidative stress markers
The concentrations of superoxide dismutase (SOD) and malondialdehyde (MDA) in cardiomyocytes were measured using corresponding commercial kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) in compliance with the manufacturer’s protocols.
Reactive oxygen species (ROS) detection assay
When ROS were detected, cardiomyocytes were collected and rinsed with PBS. Then, a ROS-sensitive probe 2’,7’-dichlorofluorescin diacetate (DCFDA) was diluted in PBS at a final concentration of 20 μM. The probe solution was used to treat cardiomyocytes. After culturing 45 min at 37°C, the fluorescence intensities of cells were measured using Varioskan Flash Multimode Reader (Thermo Fisher Scientific, Waltham, MA, USA) at an excitation/emission wavelength of 485/535 nm.
Nrf2/ARE luciferase reporter assay
The Nrf2/ARE luciferase reporter assay was performed using a pGL4.37[luc2P/ARE/Hygro] plasmid that contained ARE sites upstream of the luciferase gene. Cardiomyocytes were transfected with pGL4.37[luc2P/ARE/Hygro] plasmids, Renilla luciferase plasmids and SMS2 siRNA, cultivated for 48 h and then subjected to hypoxic conditions. The Dual-Glo Luciferase Assay System (Promega, Madison, WI, USA) was employed to quantify luciferase activity within cells.
Statistical analysis
Quantitative results were shown as means ± standard deviation. The significance of comparisons was calculated using either the Student’s t-test or one-way analysis of variance followed by Tukey’s post-hoc test. Values of P < 0.05 were considered statistically significant.
Results
SMS2 expression was elevated in cardiomyocytes following hypoxia exposure
To evaluate whether SMS2 is associated with hypoxic injury in cardiomyocytes, we investigated changes in SMS2 expression level in response to exposure to hypoxia. Results showed that mRNA expression of SM2 markedly increased following hypoxia exposure (Figure 1A). Moreover, hypoxia caused a marked increase in SMS2 protein expression in cardiomyocytes (Figure 1B and C). These data imply that elevated SMS2 expression may contribute to hypoxic injury in cardiomyocytes.
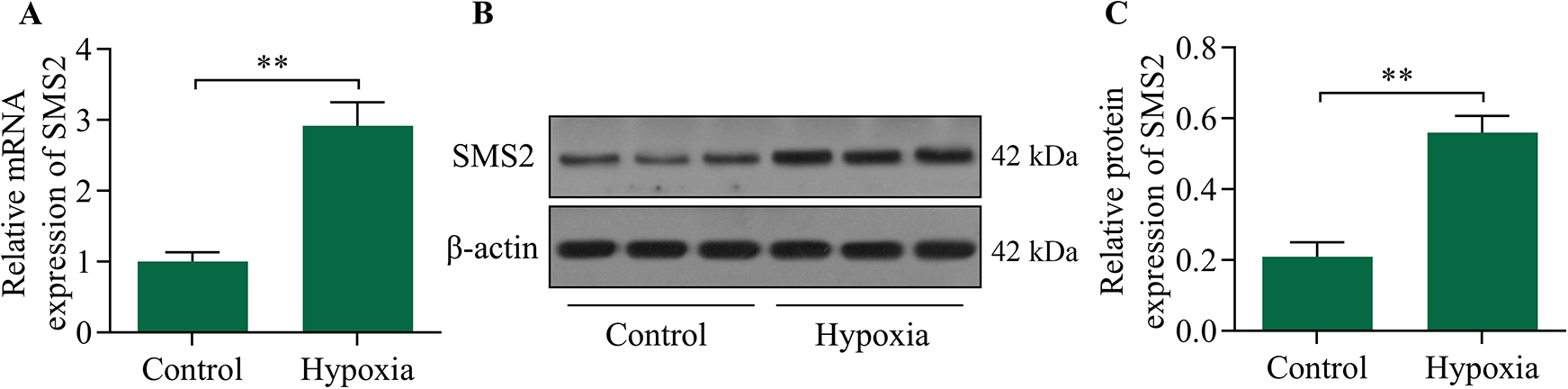
The effect of exposure to hypoxia on SMS2 expression in cardiomyocytes. Cardiomyocytes were subjected to hypoxic conditions for 24 h, and (A) expression levels of SMS2 mRNA was quantified via RT-qPCR. Levels of SMS2 proteins (B, C) were determined via Western blot. **p < 0.01.
Inhibition of SMS2 alleviated hypoxic injury in cardiomyocytes in vitro
To determine the specific role of SMS2 in the regulation of hypoxic injury in cardiomyocytes, we investigated the effect of SMS2 inhibition in cardiomyocytes exposed to hypoxic conditions. Cardiomyocytes were transfected with SMS2 siRNA to silence SMS2 expression and the downregulation of SMS2 was validated via RT-qPCR and Western blot (Figure 2A–C). The viability of cardiomyocytes decreased as a result of exposure to hypoxic conditions, and the effect was restored by knockdown of SMS2 (Figure 2D). Hypoxia-induced increases in caspase-3 activity was markedly downregulated by knockdown of SMS2 (Figure 2E). Moreover, hypoxia-induced apoptosis in cardiomyocytes was significantly reduced by knocking down expression of SMS2 (Figure 2F). In addition, SMS2 knockdown increased the levels of SOD (Figure 2G), while decreased the levels of MDA (Figure 2H) and ROS (Figure 2I) in hypoxia-exposed cardiomyocytes. Therefore, these data reveal that SMS2 inhibition ameliorates hypoxia-induced apoptosis and oxidative stress in cardiomyocytes.

Knockdown of SMS2 attenuated hypoxia-induced apoptosis and ROS generation in cardiomyocytes. Cardiomyocytes were transfected with SMS2 siRNA or negative control (NC) siRNA for 48 h and subjected to hypoxic conditions. (A) Relative SMS2 mRNA expression was assessed via RT-qPCR. (B, C) SMS2 protein expression levels were determined via Western blot. (D) The effect of SMS2 inhibition on cardiomyocyte viability was determined using an MTT assay. (E) The effect of SMS2 inhibition on caspase-3 activity was measured via a caspase-3 activity assay. (F) The effect of SMS2 inhibiting on apoptosis in cardiomyocytes was assessed using an annexin V-FITC/PI apoptotic assay. The effect of SMS2 inhibiting on levels of (G) SOD and (H) MDA was measured via commercial kits. (I) The effect of SMS2 inhibition on ROS levels in cardiomyocytes was monitored using a ROS detection assay. **p < 0.01 and ***p < 0.001.
SMS2 overexpression exacerbated hypoxic cardiomyocyte injury in vitro
To validate the role of SMS2 in the regulation of hypoxic injury in cardiomyocytes, we performed a gain-of-function experiment to assess the role of SMS2 in hypoxia-exposed cardiomyocytes. Transfection of SMS2 expression vectors into cardiomyocytes significantly upregulated SMS2 expression in hypoxia-exposed cardiomyocytes (Figure 3A and B). A viability detection assay revealed that SMS2 overexpression markedly decreased the viability of hypoxia-exposed cardiomyocytes (Figure 3C). Moreover, hypoxia-induced apoptosis (Figure 3D and E) and oxidative stress (Figure 3F–H) were significantly exacerbated by SMS2 overexpression. Thus, these results confirm that SMS2 overexpression exacerbates hypoxia-induced cardiomyocyte injury.
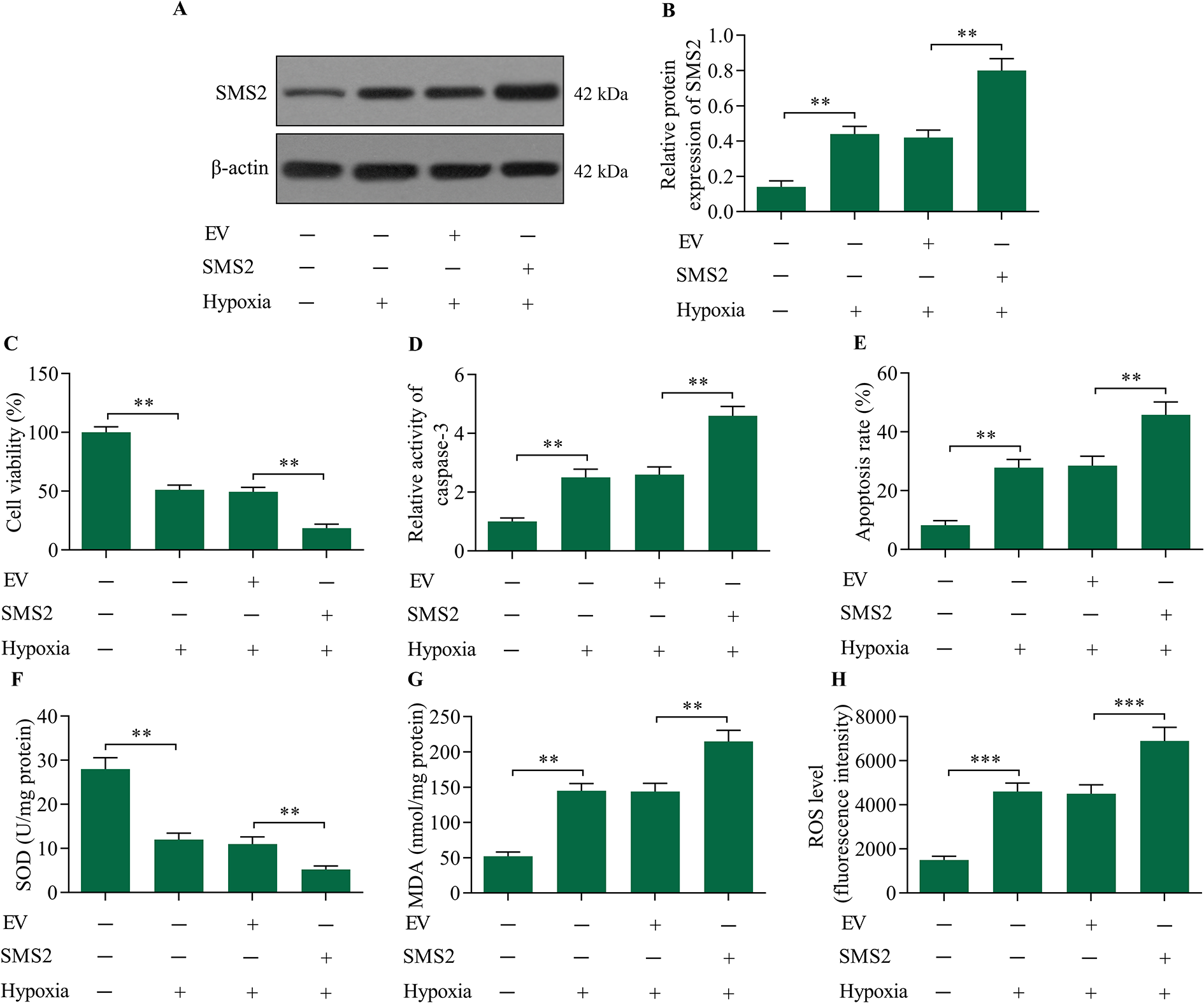
Cardiomyocytes overexpressing SMS2 were sensitive to hypoxic injury. Cardiomyocytes were transfected with either an SMS2 expression vector or empty vector (EV) control for 48 h and subsequently exposed to hypoxia. (A, B) SMS2 protein expression levels were examined via Western blot. (C) The effect of SMS2 overexpression on cardiomyocyte viability was measured using an MTT assay. (D) The effect of SMS2 overexpression on caspase-3 activity was assessed via a caspase-3 activity assay. (E) The effect of SMS2 overexpression on cardiomyocyte apoptosis was assessed using an annexin V-FITC/PI apoptotic assay. The effect of SMS2 overexpression on levels of (F) SOD and (G) MDA was detected via commercial kits. (H) The effect of SMS2 overexpression on ROS levels in cardiomyocytes was evaluated using a ROS detection assay. **p < 0.01 and ***p < 0.001.
Inhibition of SMS2 enhanced hypoxia-induced Nrf2 activation
To elucidate the molecular mechanism of the modulation of hypoxia-induced cardiomyocyte injury by SMS2, we investigated the regulatory effect of SMS2 on Nrf2 signaling. Results showed that inhibition of SMS2 significantly enhanced nuclear expression of Nrf2 (Figure 4A and B), and promoted Nrf2/ARE activation (Figure 4C). In contrast, SMS2 overexpression downregulated the activation of Nrf2/ARE signaling (Figure 4D–F). Collectively, these findings suggest that SMS2 participates in the regulation of Nrf2 signaling in hypoxia-exposed cardiomyocytes.

SMS2 inhibition enhanced hypoxia-induced Nrf2 activation. (A, B) The effect of SMS2 inhibition on the nuclear expression of Nrf2 was determined via Western blot. Histone H3 served as a loading control. (C) The effect of SMS2 inhibition on Nrf2/ARE transcriptional activity was assessed via luciferase reporter assay. (D, E) The effect of SMS2 overexpression on Nrf2 nuclear expression was determined by Western blot. (F) The effect of SMS2 overexpression on Nrf2/ARE transcriptional activity was monitored using a luciferase reporter assay. *p < 0.05 and **p < 0.01.
SMS2 inhibition enhanced Nrf2 activation through modulation of GSK-3β
To further investigate the molecular mechanism by which SMS2 regulates Nrf2, we assessed the effects of SMS2 expression on GSK-3β, a crucial regulator of Nrf2. Our data showed that SMS2 inhibition markedly enhanced phosphorylation of GSK-3β, while SMS2 overexpression reduced levels of GSK-3β phosphorylation (Figure 5A). To confirm whether SMS2 modulates Nrf2 via GSK-3β, we assessed the effect of GSK-3β inhibition on SMS2-mediated Nrf2 signaling. Data revealed that GSK-3β inhibition enhanced Nrf2 activation, and reversed SMS2 overexpression-mediated suppressive effects on Nrf2 signaling (Figure 5C). Moreover, SMS2 overexpression-mediated promotion of hypoxia-induced apoptosis and ROS production was markedly reduced by GSK-3β inhibition (Figure 5D and E). Collectively, these data indicate that SMS2 modulates Nrf2 activation via GSK-3β.
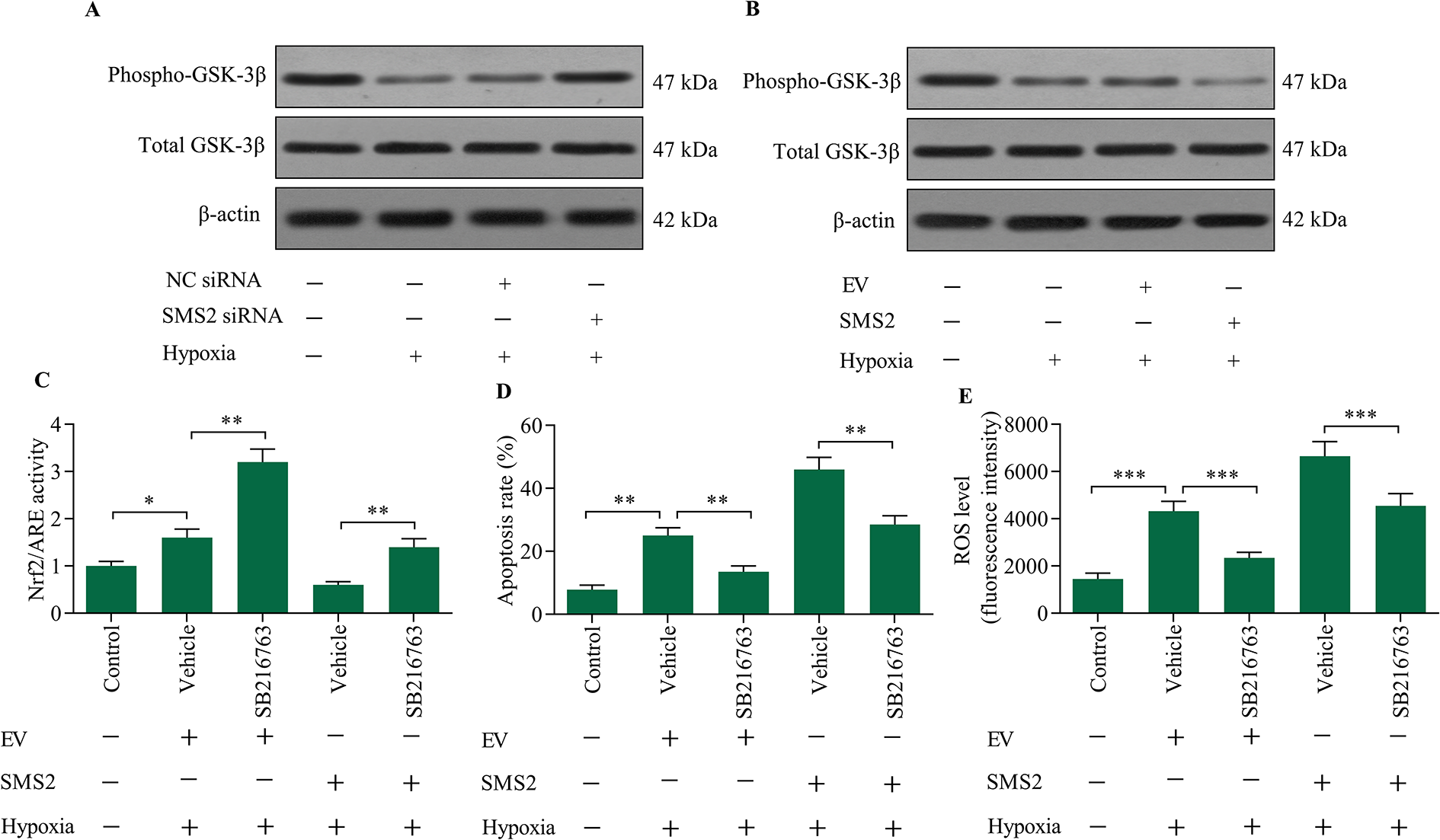
GSK-3β inhibition reversed SMS2 overexpression-mediated effects on Nrf2 signaling. The effect of SMS2 (A) inhibition or (B) overexpression on levels of phospho-GSK-3β was determined via Western blot. Cardiomyocytes were transfected with SMS2 expression vectors for 48 h in the presence of 10 μM of SB216763, a GSK-3β inhibitor, prior to hypoxia exposure. (C) The effect of GSK-3β inhibition on Nrf2 activation was monitored using a luciferase reporter assay. (D) The effect of GSK-3β inhibition on hypoxia-induced apoptosis was measured using an apoptosis assay. (E) The effect of GSK-3β inhibition on hypoxia-induced ROS generation was assessed using a ROS detection assay. *p < 0.05, **p < 0.01 and ***p < 0.001.
Nrf2 inhibition reversed SMS2 knockdown-mediated cardioprotection against hypoxic injury
To verify whether SMS2 inhibition exerts cardioprotective effects via Nrf2 activation, we further evaluated the effect of Nrf2 inhibition on SMS2 knockdown-mediated hypoxic injury in cardiomyocytes. A Nrf2 inhibitor, ML385, was used to inhibit Nrf2 activation in hypoxia-exposed cardiomyocytes transfected with and without SMS2 siRNA (Figure 6A). Nrf2 inhibition exacerbated hypoxic injury, and reversed SMS2 knockdown-induced protective effect against hypoxia-induced apoptosis and oxidative stress (Figure 6B and C). These data confirm that SMS2 inhibition protects against hypoxic injury in cardiomyocytes through enhancing Nrf2 activation.
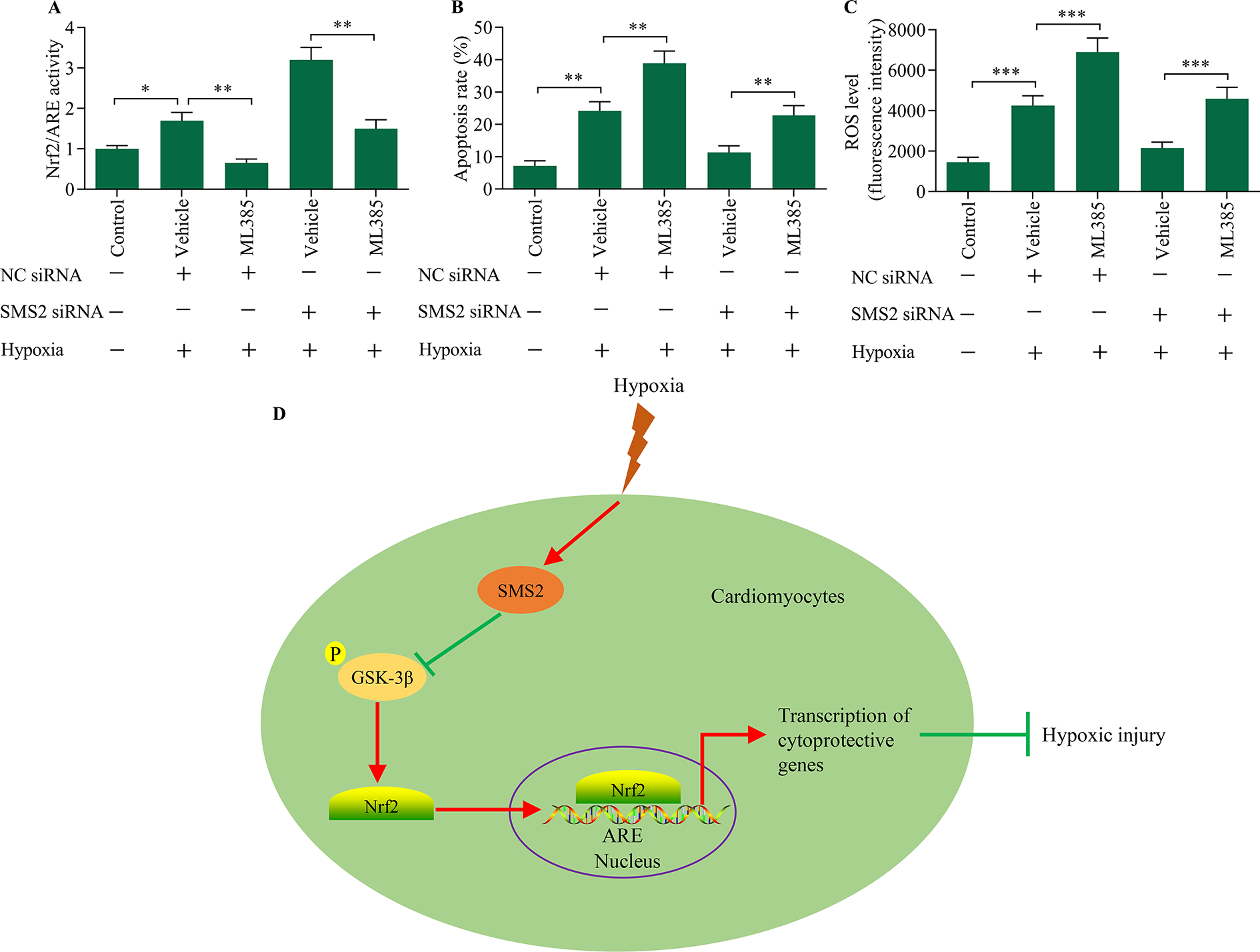
Nrf2 inhibition reversed SMS2 knockdown-mediated cardioprotective effects. Cardiomyocytes were transfected with SMS2 siRNA for 48 h in the presence of 5 μM of ML385, a Nrf2 inhibitor, prior to hypoxia exposure. (A) The effect of the Nrf2 inhibitor on Nrf2/ARE transcriptional activity was monitored using a luciferase reporter assay. (B) The effect of Nrf2 inhibition on hypoxia-induced apoptosis was measured via apoptosis assay. (C) The effect of Nrf2 inhibition on hypoxia-induced ROS generation was monitored using a ROS detection assay. *p < 0.05, **p < 0.01 and ***p < 0.001. (D) Schematic representation of SMS-mediated GSK-3β/Nrf2 signaling in the regulation of hypoxic cardiomyocyte injury.
Discussion
Our study revealed an important role for SMS2 in the regulation of hypoxic cardiomyocyte injury. Our results revealed that inhibition of SMS2 suppressed hypoxia-induced apoptosis and ROS generation in cardiomyocytes, which indicated that inhibition of SMS2 was cardioprotective. Moreover, we found that SMS2 inhibition-mediated cardioprotective effects depended on Nrf2 antioxidant signaling. Our study implies that SMS2 inhibition may contribute to Nrf2/ARE activation via inactivation of GSK-3β (Figure 6D). Therefore, our study highlights a potential role of SMS2/GSK-3β/Nrf2/ARE axis in the modulation of hypoxic injury in cardiomyocytes.
SMS2 has been suggested to be a critical regulator of cellular apoptosis and tissue injury. Overexpression of SMS2 promoted apoptosis in hepatoblastoma cells induced by cisplatin. 26 SMS2 overexpression enhanced apoptosis in endothelial cells exposed to hydrogen peroxide. 13 Downregulation of SMS2 restricted the progression of lung injury. 11,27 Moreover, inhibition of SMS2 alleviated ischemic injury within the brain. 14 To date, little is known regarding the role of SMS2 in ischemic heart injury. Herein, our results demonstrated that SMS2 plays a role in regulating hypoxic injury in cardiomyocytes, an in vitro model of ischemic myocardial injury. We determined that SMS2 expression was induced by hypoxia and knockdown of SMS2 improved cell viability and decreased apoptosis in hypoxia-exposed cardiomyocytes. However, overexpression of SMS2 upregulated hypoxia-evoked apoptosis in cardiomyocytes, indicating that hypoxia-induced SMS2 expression contributes to hypoxic injury in cardiomyocytes.
SMS2 plays an important role in the regulation of cellular oxidative stress injury. SMS2 expression was elevated in response to oxidative stress in endothelial cells. 13 Overexpression of SMS2 decreased the production of superoxide dismutase, a cellular antioxidant. 28 Moreover, inhibition of SMS2 alleviated oxidative stress-induced cellular apoptosis. 13,28 In line with these findings, we found that SMS2 overexpression aggravated hypoxia-induced oxidative injury in cardiomyocytes, while knockdown of SMS2 exhibited a remarkable antioxidative function. Therefore, our study indicates that SMS2 is involved in regulating hypoxia-induced oxidative stress in cardiomyocytes.
SMS2 acts as a key factor capable of affecting signal transduction of multiple signaling pathways, such as nuclear factor-κB, mitogen activated protein kinase, signal transducer and activator of transcription 3, and Wnt/β-catenin signaling. 28 –30 Interestingly, in this study, we found that SMS2 participated in the modulation of Nrf2 signaling. Such action of SMS2 was related to its regulatory effect on GSK-3β. We found that SMS2 overexpression promoted dephosphorylation of GSK-3β, which leads to GSK-3β activation. Indeed, SMS2 has previously been reported to play a role in regulating GSK-3β phosphorylation. 31 Activation of GSK-3β results in Nrf2 degradation, which can block activation of Nrf2/ARE signaling. Our results showed that suppression of GSK-3β expression markedly reversed SMS2 overexpression-evoked inhibition of Nrf2/ARE signaling in hypoxia-exposed cardiomyocytes. Thus, our study suggests that SMS2 mediates Nrf2/ARE signaling via modulation of GSK-3β.
In summary, the results of this study demonstrate that SMS2 participates in the modulation of hypoxic injury of cardiomyocytes by regulating the GSK-3β/Nrf2/ARE axis. We found that inhibition of SMS2 exerted cardioprotective effects by attenuating hypoxia-induced apoptosis and oxidative stress. Our study suggests that SMS2 may play a role in regulating cardiac injury related to myocardial infarction. However, the precise role of the SMS2/GSK-3β/Nrf2/ARE axis in myocardial infarction requires further in vivo investigation using animal models.
Footnotes
Author contributions
Aiping Jin: design of the work, experiment operation, drafted the article.
Haijuan Cheng: experiment operation;
Lina Xia: interpretation of data
Sha Ye: interpretation of data
Cuiling Yang: interpretation of data
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Key Research & Development Project of Shaanxi Province (2019SF-101).